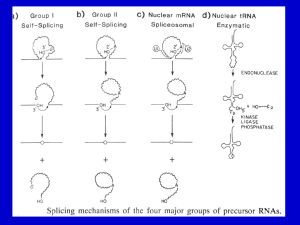
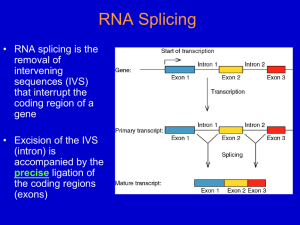
Chapter 14 Post transcriptional events I: Splicing Table of contents
advertisement

Chapter 14 Post transcriptional events I: Splicing Micrograph of human premRNA splicing factor in a cell nucleus Table of contents Genes in Pieces The Mechanism of Splicing of Nuclear mRNA Precursors Self-Splicing Introns tRNA Splicing 14.1 Genes in pieces Evidence for split genes; RNA splicing; Splicing signals 14.1.1 Evidence for split genes R-looping technique RNA products hybridize to DNA templates; Electron microscopy exams; R-looping experiments reveal introns in adenovirus Figure 14.1 R-looping experiments reveal introns In adenovirus. (a) Electron micrograph of a cloned fragment of edenovirus DNA containing the 5'-part of the late hexon gene, hybridized to mature hexon mRNA. The loops represent introns in the gene that cannot hybridize to mRNA. (b) Interpretation of the electron micrograph, showing the three intron loops (labeled A, B. and C), the hybrid (heavy red line), and the unhybridized region of DNA upstream from the gene (upper left), The fork at the lower right is due to the 3'-end o1 the mRNA, which cannot hybridize because the 3'-end of the gene is nct included. Therefore, the mRNA forms intramolecular doublestranded structures that have a forked appearance. (c) Linear arrangements of the hexon gene. showing the three short leader exons, the two introns separating them (A and B), and the long intron (C) separating the leaders from the coding exon of the hexon gene. All exons are represented by red boxes 14.1.2 RNA splicing mRNA synthesis in eukaryotes occurs I stages: 1. Synthesis primary transcription products; 2. mRNA maturation. The removal of introns in a process is called splicing Outline of splicing Figure 14.2 Outline of splicing. The introns in a gene are transcribed along with the exons (colored boxes) in the primary transcript. Then they are removed as the exons are spliced together Introns are transcribed Figure 14.3 Introns are transcribed. (a) R-looping experiment in which mouse globin mRNA precursor was hybridized to a cloned mouse β-globin gene. A smooth hybrid formed, demonstrating that the introns are represented in the mRNA precursor. (b) Similar R-looping experiment in which mature mouse globin mRNA was used. Here, the large intron in the gene looped out, showing that this intron was no longer present in the mRNA. The small intron was not detected in this experiment. In the interpretive drawings, the dotted black lines represent RNA and the solid red lines represent DNA 14.1.3 Splicing signals Exon/GU-intron-AG/exon Yeast: 5’-AG/GUAAGU-intron-YNCURAC-YNNAG/G-3’ Mammals: 5’-/GUAUGU-intron_UACUAAC-YAG/-3’ 14.2 Mechanism of splicing of nuclear mRNA precursors A branched intermediate; A signal at the branch Splicesomes Splicosome assembly 14.2.1 A branched intermediate Simplified mechanism of nuclear mRNA precursor splicing Figure 14.4 Simplified mechanism of nuclear mRNA precursor splicing, in step 1, the 2' hydroxyl group of an adenine nucleotide within the intron attacks the phosphodiester bond linking the first exon (blue) to the intron. This attack, indicated by the dotted arrow at top, breaks the bond between exon 1 and intron, yielding the free exon 1 and the lariat-shaped intronexon 2 intermediate, with the GU at the 5'-end of the intron linked through a phosphodiester bond to the branchpoint A. The lariat is a consequence of the internal attack of one part of the RNA precursor on another part of the same molecule. At right in parentheses is the branch-point showing that the adenine nucleotide is involved in phosophdiester bonds through its 2’, 3’, and 5' hydroxyl groups. In step 2, the free 3' hydroxyl group on exon 1 attacks the phosphodiester bond between the intron and exon 2. This yields the spliced exon 1 -exon 2 product and the lariatshaped intron. Note that the phosphate (red) at the 5'-end of exon 2 becomes the phosphate linking the two exons in the spliced product. Anomalous electrophoretic behavior of spliced-out intron Figure 14.5 Anomalous electrophoretic behavior of spliced-out intron. (a) Splicing substrate. Sharp and colleagues started with a cloned fragment from the adenovirus major late region containing leader exons 1 (blue) and 2 (yellow), all of intron 1 and part of intron 2, They made a deletion in intron 1, yielding a shortened intron only 231 bp long. Finally, they transcribed this construct in vitro to produce a labeled splicing substrate. (b) Electrophoresis. Sharp, and colleagues incubated the labeled splicing substrata from panel (a) with HeLa nuclear extract, then electrophoresed the resulting RNAs on (b), a 4%, or (c) a 10% polyacrylamide gel. Lane 1 and lanes 3-5 contained ATP: lane 2 did not. Lane 3 contained 2 μL of control serum; lanes 4 and 5 contained 1 and 2 μL , respectively, of an antiserum directed against an RNAprotein component required for splicing (see page 424, on snurps). Under splicing conditions (lanes 1.3. and 4), a band cot responding to the liberated intron was observed below the precursor bend in the 4% gel. but above the precursor band in the 10% gel. Time course of intermediate and liberated intron appearance Figure 14.6 Time course of intermediate and llberated intron appearance. (a) Electrophoresis. Sharp and colleagues carried out splicing reactions as described in Figure 14.5 and electrophoresed Ihe products after various limes, indicated at top, on a 10% polyacrylamide gel. The products are identified at left. The top band contained the intron-exon 2 intermediate. The next band contained the intron. Both these RNAs were lariat-shaped, as suggested by their anomalously low electrophoretic mobilities. The next band contained the precursor. The botlom two bands contained two forms of the spliced exons: Ihe upper one was still attached to the piece of intron 2. and the lower one seemed to lack Ihat extra RNA. (b) Graphic presentation. Sharp and colleagues plotted the intensities of each band from panel (a) to show the accumulation of each RNA species as a function of time. Mechanism of RNase T1 Figure 14.7 Mechanism of RNase T1. RNase T1 cuts RNA as follows: (a) The RNase cleaves the bond between the phosphate attached to the 3‘ hydroxyl of a guanine nucleotide and the 5' hydroxyl of the next nucleotide, generating a cyclic 2', 3'-phosphate intermediate. (b) The cyclic intermediate opens up, yielding an oligonucleotide ending in a guanosine 3'-phosphate. Nearest neighbor analysis Figure 14.8 Strategy of an experiment to trace the origin of the phosphate between the two exons in a spliced transcript. (a) Nearest-neighbor analysis. This lechnique is not the one Sharp and colleagues used, but it embodies the same principle. First, we label RNA by synthesizing it in the presence of an α-32Plabeled nucleotide. CTP in this case, The lathed phosphate (red) is incorporated between the C and its nearest neighbor on the 5’ side, a U in this case. Next. we hydrolyze the labeled RNA with alkali, which cleaves on thin 3' cite of every base, yielding mononucleotides. Notice that this transfers the labeled phosphate to the nearest neighbor on the 5' side, so the uracil nucleotide is now labeled instead of the cytosine nucleotide. Finally. we separate all lout nucleotides and determine the radioactivity in each. This tells us how frequently each nucleotide is C's nearest Neighbor on the 5' side. (b) Identifying the origin of the phosphate between spliced exons. Step 1: Sharp and his cilleagues labeled the splicing precursor with [α-32P] CTP, which labels the phosphate (red) between the intron and the second exon. Step 2: Our hypothesis of splicing places the labeled phosphate in the bond between the two spliced exons. The alternative scheme would involve the phosphate originally attached to the end of the first exon (blue in step 1), which would not be labeled because it entered the RNA on a GTP (the first G in the intron). Step 3: Next, Sharp and coworkers cleaved the spliced RNA with RNase A, which cuts after the pyrimidine C, yielding the oligonucleotide GpGpGpCp, where the phosphate between the last G and the C is labeled (red). Finally, they cleaved this oligonucleotide with alkali, cutting it into individual nucleotides. They analyzed these nucleotides for radioactivity and found that GMP was labeled. This indicated that the labeled phosphate at the end of the intron really did form the bridge between splice exons in the final RNA. Direct evidence for a branched nucleotide (c) Figure 14,9 Direct evidence for a branched nucleotide. (a) Sharp and colleagues digested the splicing intermediate with RNase T2. This yielded a product with a charge of -6, This is consistent with the branched structure pictured here. (b) Digestion with RNase P1 gave a product with a charge of -,4, consistent with this branched structure (c) Sharp and colleagues treated the P1 product with periodate and aniline to eliminate the nucleosides bound to the 2' and 3' phosphates of the branched nucleotide. The resulting product co-purified with adenosine 2', 3', 5'trisphosphate, verifying the presence of a branch and demonstrating that the branch occurs at an adenine nucleotide. Demonstration the the 5’-end of the intro is involved in the branch 14.2.2 A signal at the branch Figure 14.11 Demonstration of a critical signal within a yeast intron. Splicing takes place on a particle called a spliceosome. Yeast spliceosomes and mammalian spliceosomes have sedimentation coefficients of about 40S, and about 60S, respectively. 14.2.3 Spliceosomes Red: wild type Blue: mutant Figure 14.12 Yeast spliceosomes. Figure 14.13 Human spliceosomes Snurps (small nuclear RNAs, snRNAs) Figure 14.14 Splicing scheme of adenovirus E1A gene and RNase protection assay to detect each spliced product. (a) (b) Figure 14.15 Alignment of wild-type and mutant 5’ splice site with wild-type and mutant U1 snRNAs Results of RNase protection assay Figure 14.16 Results of RNase protection essay. Zhuang and Weiner tested the wild-type and mutant 5' splice sites and wild-type and mutant U1 snRNAs pictured in Figure 14.15 by transfecting HeLa cells with plasmids containing these genes, then detected splicing by RNase protection as illustrated in Figure 14.14. Lane 1, size markers, with lengths in base pairs indicated at left. Lane 2, mock-transfected cells (negative control). Lane 3, wild-type EIA gene with wild-type U1 snRNA. Signals were visible for the 13S and 12S products, but not for the 9S product, which normally does not appear until lane in infection. Lane 4, mutant hr440 with an altered 12S 5' splice site. No 12S signal was apparent. Lane 5, mutant hr440 plus mutant U1 snRNA (U1-4u). Splicing at the 12S 5' site was restored. Lane 6, mutant pm1114 with an altered 13S 5' splice site. No 13S signal was apparent. Lane 7, mutant pm1 114 plus mutant U1 snRNA (U1-6a)o Even though base pairing between 5‘ splice site and U1 snRNA was restored, no 13S splicing occurred. Figure 14.17 Two models for interaction between a yeast 5’ splice site and U6 snRNA. (a) (b) (c) Figure 14.18 Reporter genes used to detect normal and aberrant splicing. (a) (b) Figure 14.19 Test of model 1 base-paring. (a) (b) (c) Figure 14.20 Summary of a test of model II base-paring. Crosslinking of splicing intermediates with 4-thioU Figure 14.21 Crosslinking of splicing intermediates with 4-thioU. Sontheimer and Steitz incubated a labeled adenovirus splicing precursor containing 4-thioU in the second position of the intron with a nuclear splicing extract. After various periods of time (indicated at top), they removed samples, uvirradiated them to cross-link the 4-thioU to any RNAs in the neighborhood, and electrophoresed the products. Lane 1, input RNA with no incubation; lane 2, 20minute incubation with no nuclear extract (NE); lane 3, 20 minute incubation followed by no uv-irradiation; lanes 4-12, incubation for the times indicated at top; lane 13, no 4-thioU labeling; lane 14, no ATP; lane 15, EDTA added to chelate magnesium and block splicing; lane 16, a fraction clarified by high-speed ultracentrifugation was used instead of nuclear extract. The dot at left denotes a band resulting from an intramolecular cross-link. Figure 14.22 Two-dimensional electrophoresls to detect splicing complexes. Figure 14.22 Two-dimensional electrophoresls to detect splicing complexes. (a) Complexes involving U 1, U2, U4, and U5. Wassarman and Steitz formed splicing complexes using a biotin-labeled splicing precursor, cross-linked RNAs in the complexes with psoralen, and isolated snRNP-containing complexes by first precipitating with an antim3G antibody (which recognizes all snRNPs except U6). then precipitating with streptavidin beads, which recognize the splicing procurer. Next they end-labeled the RNAs in the complexes with [32P]pCp and RNA ligase, then electrophoresed these complexes in the horizontal dimension as shown at top right. They dissociated the complexes with ultraviolet light (254 nm) and electrophpresed the RNAs in the vertical dimension to identify each RNA in each complex. The marker positions at right identify the individual RNA species in each complex, and the mixtures of RNAs in the complexes are suggested at bottom. Sometimes. two or more complexes co-purified This was the case with U2/U4/U6, a U1 internally linked molecule, and the U2/lariat complex. (b) Complexes containing U6. Since U6 does not have a m3G cap, Wassarman and Steitz isolated complexes containing this snRNP using a biotin-labeled U6-specific oligonucleotide and avidin beads. Other conditions were as in panel (a). The marker positions at right identify both splicing substrate and lariat, as well as U2, as components of U6-containing complexes. U6 itseff is not detectable in the first two of these complexes, but we know it is there, since these complexes were selected by a U6-specific probe Figure 14.23 Base paring between yeast U2 and yeast branch point sequence. Figure 14.24 Growth of A257 (panel a) and A256 (penel b) mutants on Hol medium in the presence of wild-type and suppressor mutant U2. The abbreviations under each patch of cells denote the nature of the U2 added, if any: UT, untransformed (no U2 added); WT, wild-type U2; U36, U2 with mutation that restores base pairing with A257; U37, U2 with mutation that restores base pairing with A256; LP, a colony that lost its U2 plasmid, The positive control in each plate (+) contained a wild-typa fusion gene and no extra U2. The negative control in each plate contained no fusion gene. Figure 14.25 Detection of a complex between U5 and the 5' end of the second exon. Figure 14.25 Detection of a complex between U5 and the 5' end of the second exon. (a) Forming the complex. Sontheimer and Steitz placed 4-thioU in the first position of the second exon of a labeled splicing substrata and cross-linked in to whatever RNAs were nearby at various times during splicing. Then they electrophoresed the products and detected them by autoradiography. The US/intron-E2 doublet appears near the top late in the splicing process (after 30 minutes), Lane designations are as in Figure 14.21. (b) Identification of the RNAs in the complex. Sontheimer and Staitz irradiated the splicing mix after 30 minutes of splicing to form crosslinks, then incubated it with DNA oligonuclaotides complementary to U5 and other RNAs, then added RNase H to degrade any RNAs hybridized to the oligonucleotides. Finally, they electrophoresed and autoradiographed the products. The oligonucleotides (oligos) used were as follows: lanes 1 and 5, no oligo; lane 2, anti-exon 1 oligo; lane 3, anti-intron oligo; Lane 4, anti-exon 2 oligo; lane 6, anti-US oligo. The anti-intron, anti-exon 2, and anti-US oligos all destroyed the complex, indicating that the complex is composed of the intron, second exon, and US. Figure 14.26 Identification of snRNP bases cross-linked to 4-thioU In various positions in the slicing substrate. Figure 14.26 Identification of snRNP bases cross-linked to 4-thioU In various positions in the slicing substrate. Sontheimer and Steitz used primer extension to map the bases in U5 and U6 cross-linked to 4-thioU in the following positions: the last base in the first exert (Ad5-1, panels a and b); the second base in the intron (Ad5+2, panel c); or the first base in the second exert (Ada+l, panel d). They formed cross-linked complexes with these RNAS, then excised the complexes from the electrophoresis gels and added primers specific for either U5 or U6, and performed primer extension analysis. The first four lanes in panels (a-c) and lanes 5-8 in panel (d) are sequencing lanes using the same primer as in the primer extension assays The lanes marked "blank" are control sequencing lanes with no template. The experimental lanes are lanes 6 in panels (a and b), lanes 6 and 8 in panel (c), and lane 1 in panel (d). These are the results of primer extension with: the US/splicing precursor complex (US/pro, panel a); the U5/exon 1 complex (U5/E1, panel b); the U6/inntron-exon 2 complex (U6/intron-E2, panel c), and the U6/intron complex, panel (c); and the U5/intron-exon 2 complex (U5/intron-E2, panel d). The other lanes are controls as follows; "no substrate," substrate was omitted from the reaction mix, then a slice of gel was cut out from the position where complex would be if substrate wore included; "UV RNA," total RNA from an extract lacking substrate: "pre-mRNA," uncross-linked substrate. The c~ss-linked bases in the snRNPs are marked with dots at the Left of each panel. Figure 14.27 Summary of U5 and U6 interactions with the splicing substrates revealed by 4-thioU crosslinking. Figure 14.28 Postulated base paring between U4 and U6. Figure 14.29 A model to compare the active center of a spliceosome to the active center of a group II intron. 14.2.5 Spliceosome assembly Figure 14.30 Kinetics of association of spliceosomal snRNPs with pre-mRNA. Effect of inactivation of U1 or U2 on assembly of the spliceosome Figure 14.31 Effect of Inactivation of U1 or U2 on assembly of the spliceosome. Figure 14.31 Effect of Inactivation of U1 or U2 on assembly of the spliceosome. Ruby and Abelson inactivated either U1 or U2 by incubation with RNase H and a DNA oligonucleotide complementary to a key part of either snRNA. Lanes 11-15 show the patterns of labeled snRNAs in an extract after treating with RNase H and: no oligonucleotide (No); an anti-U1 oligonucleotide (U1); an anti-U2 oligonucleotide (U2); or an anti-T7 oligonucleotide (1"7). The latter sewed as a second negative control. Treatment with RNase H and anti-U1 led to essentially complete conversion to a truncated form that electrophoresed slightly faster than the parent RNA. Treatment with RNase H and anti-U2 led to near-elimination of full-size U2, and appearance of a small amount of truncated U2. Lanes 1-10 show the results of spliceosome assembly experiments, as described in Figure 14.30, under the following conditions, as indicated at top: C303/305 pre-mRNA, or no prs-rnRNA; extracts treated with RNase H and no oltgonucleotide, anti-U1, anti-U2, or anti-T7 oligonucleotides; and with or without ATP. Inactivating U 1 prevented binding of U1, U2, and U5. Inactivating U2 prevented binding of U2 and U5. Figure 14.32 The spliceosome cycle. Figure 14.29 Structure of U1 snRNP. Stark and colleagues used single-particle electron cryomicroscopy to obtain this stereo model of the snRNP structure. The major protrusions, including the U1-A and 70K proteins, from the central Sm "doughnut" are labeled. Commitment of the human β-globin premRNA Figure 14.33 Commitment of the human β-globin pre-mRNA. Figure 14.33 Commitment of the human β-globin pre-mRNA. (a) Commitment competition assay with general splice site competitor RNAs. Xiang-Dong Fu used a competition assay for commitment as follows. He incubated a labeled human β-globin preomRNA with or without SC35, as indicated at top (+ and -, respectively). Then he added a nuclear extract with or without a competitor RNA, as indicated at top. No competitor is indicated by (-). C1 and C2 are non-specific RNAs that should not interfere with splicing, RNAs containing 5' and 3' splice sites are indicated as 5'SS and 3'SS, respectively. After allowing two hours for splicing, Fu electrephoresed the labeled RNAs and autoradiogrephed the gel. The positions of pre-mRNA and mature mRNA are indicated at right. SC35 caused commitment. (b) Commitment assay with specific competitor. Fu re-ran the competition assay with the human β-globin pre-mRNA as competitor. Again, SC35 caused commitment, especially if added before the competitor (prsincubation). (c) Conditions favoring commitment. Fu ran the commitment assay again under the conditions listed at top. Figure 14.34 Commitment activities of several RNA-binding proteins with the human β-globin pre-mRNA. Fu ran the commitment assay with two different concentrations of the seven different RNA-binding SR proteins listed at top. Lane 1 contained no competitor and no SR protein; lane 2 contained competitor, but no SR protein; lanes 3 and 4 included competitor, but no preincubation with SC35. All other lanes included competitor and preincubation with an SR protein as usual. The positions of the pre-mRNA and mature mRNA are shown at right. Figure 14.35 Commitment activities of several RNA-bindlng proteins with either the β -globin pre-mRNA or the HIV tat pre-mRNA. (a) Effect of SF2/ASF on commitment with tat pre-mRNA. Fu ran the commitment assay with the concentrations of the SF2/ASF shown at top, and either without (lanes 1-3) or with (lanes 4 and 5) preincubation with the splicing factor. Comparing lanes 5 and 3 gives the clearest view of the effect of SF2/ASF. (b) Specificity of SR proteins. Fu tested two different concentrations of the seven different SR proteins listed at top for commitment with the tat pre-mRNA. Lanes 1 and 2 contained human β globin pre-mRNA with and without preincubation with SC35. All other lanes contained tat premRNA. The positions of the β -globin pre-/and mature mRNA are indicated at left ; the positions of the tat pre-/and mature mRNA are indicated at right. The stars denote bands resulting from artifactual tat pre-mRNA degradation. (a) (b) Figure 14.36 principle of the yeast two-hybrid assay. (c) Figure 14.37 Yeast two-hybrid assays for interactions between BBP and other proteins. Figure 14.38 Summary of intronbridging protein-protein interaction in yeast and mammals. Role of the RNA polymerase II CTD The CTD of the Rpb1 subunit of RNA polymerase II stimulates splicing of substrates that use exon definition, but not those that use intron definition, to prepare the substrate for splicing. The CTD binds to splicing factors and could therefore assemble the factors at the ends of exons to set them off for splicing. CTD-GST stimulates splicing in vitro, by Zeng et al. Effect of CTD-GST on splicing using exon or intron definition. By Zeng et al. Model for participation of CTD in exon definition. Figure 14.39 Alternative splicing pattern in the mouse immunoglobulin μ heavy chain gene. Figure 14.40 Alternative splicing cascade in Drosophila sex determination. Commitment assay for femalespecific splicing of dsx pre-mRNA Figure 14.41 Commitment assay for female-specific splicing of dax pre-mRNA. Tian and Maniatis assayed for the ability of various SR proteins to complement Tra and Tra-2 in an in vitro dax splicing essay. Lane I contained no complementing protein, Lane 2 contained a mixture of SR proteins precipitated by ammonium sulfate (AS), Lanes 3-14 contained various amounts of the SR proteins indicated at the top of the lanes. Lane 15 is another negative control identical to lane 1. Lane 16 contained the highest amount of recombinant SC35, as in lane 11. Lanes 17-20 contained the purified nonrecombinant SR proteins indicated at the top of each lane. The electrophoretic mobilities of the splicing substrate (top band) and the spliced product (bottom band) are indicated between the two autoradiographs. Figure 14.42 In vitro transcription of plasmlds containing the 26S rRNA intron yields excised introns. Cech and colleagues used E.coli RNA polymarase to transcribe two different plasmids containing the rRNA intron. The reaction mix contained labeled nucleotides to label the products. These workers electrophoresed the labeled RNA products and visualized them by autorediography. Lane 1, transcripts from EcoRI-cleaved pIVS13; lane 2, linear and circular intron markers; lane 3, transcripts from EcoRIcleaved plVS11; lane 4, transcripts from lane 3 incubated for 30 minutes under conditions that favor circularization of the intron (higher temperature, MgCl2, and ammonium sulfate concentration); lane 5, transcripts from supercoiled pIVS11 ; lane 6, transcripts from lane 5 treated as in lane 4, The large RNAs in lane 5 are indistinct because the template was not linearized, so there was no definite stop-point for transcription. At left are the positions of the circular and linear introns, as well as the linear intron after losing 15 nt from its 5'end. Figure 14.43 The splicing reaction can be separated from the transcription reaction. (a) Transcription. Cech and colleagues transcribed plasmid plVS11 under conditions that inhibited splicing (presence of polyamines), then electrophoresed the labeled products. Four bands (designated a-d at left) appeared; a and b were difficult to distinguish in this experiment. (b) Splicing. Cech and coworkers took RNA from the origin and bands a-d from panel (a) and either electrophoresed them directly (lanes -) or incubated them under splicing conditions (no polyamines) before electrophoresis (lanes marked +). The nature of the RNA (odgin or band a-d) is indicated at top. The RNA at the origin end in band a of panel (a) yielded no intron, while the RNAs from bands b-d did yield intron, but only under splicing conditions. Figure 14.44 Autocyclizatlon of the linear intron. Cech and colleagues isolated labeled linear intron, either from Tetrahyrnena nuclei or from an in vitro splicing reaction, by electrophoresis, treated it in various ways, then re-electrophoresed the products. Lane 1, untreated linear intron from nuclei; lane 2, linear intron from nuclei incubated under cyclization conditions (high temperature and high MgCl2 and salt concentrations); lane 3, circular intron marker; lane 4, linear intron produced by transcription of pIVS11 and splicing the RNA in vitro," lane 5, linear intron from lane 4 incubated under splicing conditions. The minor band just above the linear intron in lanes 4 and 5 is an unrelated transcript from the plasmid. Positions of circular (C), linear (L) introns are indicated at left and right. The position of the linear intron lacking 15 nt at its 5'-end (L-15) is indicated at left. 14.3 Self splicing RNAs Group I introns Group II introns Figure 14.45 Demonstration of exon ligation. Figure 14.46 Addition of GMP to the 5' end of the excised intron. (a) Radioactive GTP labels the intron during splicing. Cech and coworkem transcribed plasmid plVS11 under non-splicing conditions with no labeled nucleotides. They isolated this unlabeled 26S rRNA precursor and incubated it under splicing conditions in the presence of [c~-32P]GTP. Then they chromatographed the products on Sephadex G-50, electrophoresed the column fractions, and autoradiographed the gel. Lanes 1-4 are successive fractions from the Sephadex column. Lane 5 is a linear intron marker. Lanes 2 and 3 contain the bulk of the linear intron, and it is labeled, indicating that it had incorporated a labeled guanine nucleotide. (b) Sequence of the labeled intron. Cech and colleagues used an enzymatic method to sequence the RNA. They cut it with base (OH-), which cuts after every base; RNase Phy M, which cuts after A and U; RNase U2, which cuts after A; and RNase T1, which cuts after G. Treatment of each RNA sample is indicated at top. The deduced sequence is given at left. Note the 5'-G at bottom. Figure 14.47 Self-splicing of Tetrahymena rRNA precursor. Figure 14.48 Fate of the linear Intron. We begin with the linear intron originally excised from the 26S rRNA precursor. This can be cyclized in two ways: In reaction 1, (green arrows), the 3' terminal G attacks the bond between U-15 and A-16, removing a 15 nt fragment and giving a circular intron (C-15). In the alternative reaction (2, blue arrows), the terminal G attacks 4 nt farther into the intron, removing a 19 nt tragment and leaving a smaller circular intron (C-19). Reaction 3, C-15 can open up at the same bond that closed the circle, yielding a linear intron (L-15). Reaction 4, the terminal G of L-15 can attack the bond between the first two Us, yielding the circular intron C-19. Reaction 5, C-19 opens up to yield the linear intron L-19. Figure 14.49 Two views of GMP held in a pocket of the 26S rRNA intron. (a) A cross-eyed stereogram that can be viewed in three dimensions by crossing the eyes until the two images merge. Carbon atoms of RNA, green; carbon atoms of G, yellow; phosphorus, lavender. Other atoms are standard colors. (b) Space-filling model. Colors are as in part (a). Figure 14.50 Lariat-shaped splicing intermediate and product. Electron micrograph of intermediate and product of yeast mitochondrial rRNA splicing provides direct visual evidence for lariats. (Top) Two lariat-shaped introns, (Bottom) Two lariat-shaped intermediates containing an intron plus the second exon. 14.4 tRNA splicing Figure 14.51 Consensus tertiary structure of yeast tRNA precursors. Figure 14.53 Mechanism of tRNA splicing.