Glycogen storage diseases
advertisement

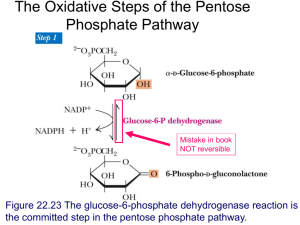
PHM142 Fall 2015 Instructor: Dr. Jeffrey Henderson Glycogen Storage Disease Mechanisms Amandeep Kaur Dhaliwal, Navneet Madhar, Onella Pereira and Superna Ramesh October 6th, 2015 Overview 1. Introduction to Glycogen Storage Disease 2. Type 4: Transglucosidase Deficiency 3. McArdie’s Disease 4. von Gierke Disease 5. Tarui Disease 6. Disease Management 7. Summary Glycogen Storage Disease (GSD) ● Inability for the body to break down glycogen to free glucose ● Type I-VII and Type O ● Affects all organs, namely liver, kidneys and muscle Glycogen Synthesis •Glycogen branches are formed by the branching enzyme amylo-(1,4→1,6)-transglycosylase • Transfer of 6- or 7-residues to chain ≥ 11 residues • Polymer adds additional non-reducing end for glycogen synthase Type IV • Deficiency of enzyme -Normal amount of glycogen -Longer outer branches -Impact on storage reserve GSD Mechanism : McArdie’s Disease GSD Mechanism : von Gierke’s Disease GSD Mechanism: Tarui Disease ● Enzyme deficiency decreases the rate of conversion of fructose-6phosphate to fructose-1,6bisphosphate ● Slows breakdown of glycogen ○ Prevents normal breakdown of glucose ○ Accumulation of glycogen in muscles ○ Lack of fuel sources for muscles Disease Management ● No specific treatment ● Diet therapy beneficial in certain cases ● High protein diet to increase muscle function in patients with muscle weakness or exercise intolerance ● Monitor glucose levels ● Rest, in some cases Summary: Glycogen Storage Diseases Target Gene Enzyme Affected Type I G6PC and SLC37A4 Glucose-6-phosphatase Glucose-6-translocase Type IV GBE1 Amylo α1,4 - α1,6transglucosidase Type V PYGM Myophosphorylase Type VII PFKM Phosphofructokinase ● Management: Monitoring lifestyle (exercise and glucose) ● All diseases result in an increase in glycogen References “Glycogen Storage Diseases.” Coriell Institute for Medical Research. Accessed Sept 17, 2015 <https://catalog.coriell.org/0/Sections/Collections/NIGMS/gsd_pathway.aspx?PgId=254>“ “Glycogen storage disease type I.” Genetics Home Reference. 2015. U.S. National Library of Medicine. Accessed Sept 17, 2015 <http://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-i> “Glycogen storage disease type IV.” Genetics Home Reference. 2015. U.S. National Library of Medicine. Accessed Sept 17, 2015 <http://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-iv#diagnosis>McArdle Syndrome.” “Glycogen storage disease type V.” Genetics Home Reference. 2015. U.S. National Library of Medicine. Accessed Sept 17, 2015 <http://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-v> “Glycogen storage disease type VII.” Genetics Home Reference. 2015. U.S. National Library of Medicine. Accessed Sept 17, 2015 <http://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-vii#diagnosis> J. Berg., J. Tymoczko. & L. Stryer. “A Biochemical Understanding of Glycogen-Storage Diseases Is Possible.” Biochemistry 5th Ed. H. Freeman and Company. New York. 2002. Section 21.5.4. Accessed through NCBI <http://www.ncbi.nlm.nih.gov/books/NBK22444/> Medline Plus. 2015. U.S. National Library of Medicine. Accessed Sept 17, 2015 from <https://www.nlm.nih.gov/medlineplus/ency/article/000329.htm> R.H. Garret, C.M. Grisham, S. Andreopoulos, Willmore and I.E. Gallouzi, Biochemistry, First Canadian Edition, Nelson (2013); “Type Ia Glycogen Storage Disease” Medscape. Wayne E Anderson. 2015. Accessed Sept 17, 2015 <http://emedicine.medscape.com/article/119318-overview>