Pediatric Puzzler
advertisement

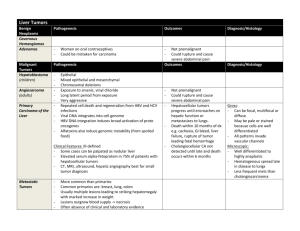
August 2007 “My baby is not right and she’s got yellow eyes” 5 week old Caucasian female who presents with lethargy, jaundice and poor weight gain. PCP referred to a gastroenterologist for rising direct hyperbilirubinemia Birth history: FT C/S for failure to progress BW 3.61 kg NICU for 6 days b/c of hypothermia and hypoglycemia sepsis w/u done was negative NICU labs: initial glucose 27 bili t/d (6dol) 15.3/1 Diet: Breastfed until DOL 8 when changed to hydrolyzed protein formula secondary to persistent elevated bili. Feeding well per mom Elimination: 10 wet diapers/day, 2 yellow-brown seedy stools/day Patient was seen by PCP for jaundice at 5 weeks. She was noted to have a bili t/d of 9.5/4.5 Patient was referred to a gastroenterologist for further workup What is direct hyperbilirubinemia exactly? Direct bilirubin >2 mg/dL Direct bilirubin > 20% of total bili Direct hyperbilirubinemia effects 1 in 2500 infants. That’s fairly common! BUT…it’s never normal! Don’t be afraid to ask… Wt 3.55 kg (10-25%) Lt 55 cm (75%) HC 39 cm (50%) T 98.7 P 160 R 36 B/P 92/58 Gen: quiet, responds to mom, no apparent distress, minimal subcutaneous fat, no dysmorphic features HEENT: mm dry, sclera icteric, OP clear CV/RESP: RRR no murmurs, CTAB Abd: soft, ND, no HSM, no masses GU: normal female Skin: capillary hemangioma in right axilla Neuro: grossly intact, + moro, +suck, +grasp Top 3 diagnoses Top 3 tests …with rationale por favor Elevated LFTs Hepatocellular injury Elevated Alk phos and GGT-- point to obstructive conditions Make sure to fractionate the bili! TORCH titers, Ucx, Sweat test, Urine reducing substances if indicated by Hx/PE Follow up on NBS! Consider liver function studies as well. 139 106 13 101 4.6 18 0.7 10.3 Alb 2.3 Bili t/d 8.9/4.2 AST 50/ALT 22/ AP 385 ESR 8 (0-20) NBS- normal from the state lab 14.6 11 537 33 s 14 l 81 m3 Ammonia 58 (29-70) α 1 antitrypsin - normal UA- 1.004 pH 5; 1 WBC; 0 RBC; no RS Cultures sent BCx, UCx, CSF Cx IVF IV Antibiotics Breastfeed or formula ad lib Is and Os monitored Scans ordered Abdominal u/s Reason for order: Exclude surgical cause of extrahepatic biliary obstruction Result: NORMAL Hepatobiliary nuclear scan Reason for order: Look for patency of biliary tract R/O biliary atresia Result: NORMAL, gallbladder visualized and excretion was normal The astute T3, just joking it’s a L3, questions the UOP on rounds. She noticed that it was 6 cc/kg/hr. The resident had failed to check the baby’s Is & Os for the day. Whoops. As a group, refine your problem definition and differential Do we need any other labs/ tests? Why? Intrahepatic Cholestasis Primary Alagille Syndrome Idiopathic Neonatal Hepatitis Acquired Drugs/ Fetal Alcohol Sepsis TORCH Hep B HIV TPN Endocrine Disorders Hypothyroidism Hypopituitarism Extrahepatic Cholestasis Biliary Atresia Choledochal cyst Choledocholithiasis cyst Genetic/ Metabolic α 1 antitrypsin Cystic fibrosis Galactosemia/ Fructosemia Gaucher disease Niemann Pick Glycogen storage disease Trisomy 18 Trisomy 21 Tyrosinemia Alagille Syndrome AD genetic d/o with paucity of bile ducts. May also have PPS and vertebral defects Features: •Long nose with bulbous tip •Triangular face •Broad forehead Most common and serious cause of direct hyperbilirubinemia Destroys the intra- and extrahepatic biliary tree May also see acholic stools Should be diagnosed before 2 months--- HIDA Tx: Kasai then transplant Idiopathic neonatal hepatitis Self limited- resolves in 3-4 weeks Giant cells are seen on biopsy Alpha 1 Antitrypsin Most common genetic cause of acute and chronic liver disease in children Liver retains abnormally folded protein May also develop lung disease (later in life) Coagulopathy Fat malabsorption Ascites Encephalopathy What do we think about UOP of 6 cc/kg/hr? What could be the cause? Looking back at labs done during the hospitalization, it was noted that the serum sodium peaked at 149 mEq/L What are we worried about now? “Flavorless Urine” Characterized by: Inability to concentrate urine Polyuria Polydipsia Cause ADH deficiency- Central Lack of end organ response to ADH- Nephrogenic Thick limb of ascending loop of Henle Collecting duct Characteristics in infancy FTT Vomiting Constipation Unexplained fevers Shock (hypovolemic) Convulsions Urine Osmolality= 167 mOsml/L (>200) Serum Osmolality= 300 mOsml/L (275-295) DDAVP was given UOsm of 310 mOsml/L What type of Diabetes Insipidus is this? CENTRAL The kidney responded appropriately to DDAVP dose. DI SIADH High serum Na Low serum Na Low urine SG <1005 Low serum Cl Low urine osm 50-200 Inappropriately Low vasopressin concentrated urine Euvolemic or hypervolemic Tx: Fluid restrict Tx: DDAVP Why does this girl have central DI? What tests do we want now? MRI was done- Pituitary was completely normal! In Central DI- the signal on T1 is usually abnormal (not as bright (hyperintense) as usual) Further investigation of the pituitary was done. Why? Central DI The glucose in the NICU was low ? Cortisol deficiency ? Growth hormone deficiency Result: Serum cortisol low for age The resident went back to spend some time examining and observing the infant. She noted that the eyes seemed to “wander”. That is, the baby could not fix her gaze Why? What should you do at this point? Ophthalmology consult was obtained Impression: Bilateral optic nerve hypoplasia So what do we have now? Direct hyperbilirubinemia Central Diabetes Insipidus Cortisol deficiency Optic Nerve Hypoplasia Can we tie all these problems into one diagnosis? Septo-Optic Dysplasia (SOD) One of the most common forms of congenital growth hormone deficiency Results from incomplete development of forebrain & pituitary Must have 2 of these 3 features for a diagnosis Optic nerve hypoplasia Absent septum pellucidum/corpus callosum Pituitary endocrinopathy (deficiency) Isolated GH deficiency Multiple pituitary hormone deficiencies May also see other midline defects (ex cleft lip) MRI results widely vary Varying degrees of endocrine disturbance seen None GH TSH LH/FSH ACTH Prolactin ADH Did you know?? Hypopituitarism can lead to cholestasis Short stature Precocious/ Delayed Puberty Diabetes Insipidus (25%) Most cases are sporadic but familial forms do occur suggesting genetic defects may play an important role Viruses Teratogens Continued to have low cortisol (replaced) Became hypothyroid and was started on thyroxine Direct hyperbilirubinemia resolved with DDAVP What are your take away points? Hope you enjoyed the puzzler and learned something new!