biochem 38 [4-20
advertisement

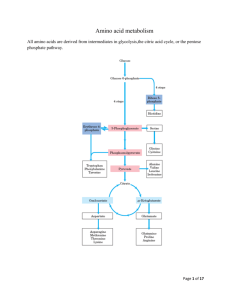
Chapter 38 Urea Cycle Learning Objectives 1. What is the major process for removing nitrogen from amino acids called? Which amino acids cannot undergo this process? Transamination is the major process which removes nitrogen from AAs Lysine and threonine cannot undergo transamination 2. What cofactor does transamination require? For what other reaction is this cofactor needed? Piridoxal phosphate (from vit. B6/pyridoxine) is required for transamination Piridoxal phosphate is also needed for glycogenolysis Piridoxal phosphate = PLP 3. What are the usual products of transamination (i.e. what is the nitrogen source)? During transanimation, α -ketoglutarate is transformed into glutamate, and another AA is converted to its corresponding α-keto acid 4. What enzyme deaminates glutamate? What is the second function of this enzyme? Glutamate dehydrogenase deaminates glutamate into α-ketoglutarate and ammonia Enzyme is reversible! It also can aminate α-ketoglutarate into glutamate 5. What is deamidation? Deamidation is the removal of nitrogen from either glutamine or asparagine How do muscle and brain form ammonium specially? Muscle and brain can produce NH4+ during the purine nucleotide cycle 6. 7. What does A vitamin B6 deficiency cause to appear in the urine? Other symptoms? xanthurenic acid appears in the urine due to the stalling of tryptophan degredation, along with microcytic anemia and dermatitis 8. How is ammonia transported to the liver from muscle? What is this process called? Ammonia is transported to the liver as alanine, after being transaminated from pyruvate and glutamate i. The reason, I think, is that muscles don’t possess the enzymes necessary to degrade AAs into glucose This is the glucose/alanine cycle 9. Where is glutamine synthetase located in the liver? Glutamine synthetase is concentrated around the portal vein, where it can prevent the escape of ammonia by incorporating it into glutamine 10. Name the five enzymes of the urea cycle in order: Carbamoyl phosphate synthetase I (CPSI) Ornithine transcarbamoylase (OTC) Arginosuccinate synthetase Arginosuccinate lyase Arginase [the names get shorter as the cycle continues!!!] [for intermediates: Ordinarily, Careless Crappers Are Also Frivolous About Urination] 11. Where do the 2 nitrogen atoms in urea come from? Ammonium (made into carbamoyl phosphate) and aspartate are the sources of nitrogen in the urea cycle i. Don’t be fooled by the production of another AA during the cycle! 12. Where is ornithine synthesized de novo when supplies run low? Ornithine aminotransferase is in the intestine 13. How does arginine stimulate the urea cycle (2)? Arginine increases the synthesis of NAG, which stimulates the action of carbamoyl phosphate synthetase i. Arginine levels will be elevated when protein has been consumed for example Since arginine is an intermediary in the urea cycle, it also increases urea production by itself 14. What is the most common defect in the urea cycle? How is it inherited? Ornithine transcarbamoylase (OTC) deficiency is the most common disorder It is X-linked 15. How are defects in the urea cycle managed? Ingesting large amounts of arginine will treat OTC, argininosuccinate synthetase, and argininosuccinate lyase deficiencies, since ornithine can be regenerated i. Nitrogen can still be excreted as argininosuccinate However, an arginase defect means that the patient cannot regenerate ornithine i. In this case, administer benzoic acid (reacts with glycine) and phenylbutyrate (reacts with glutamine)