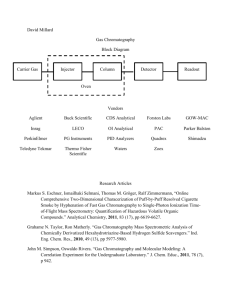
Lecture 6: Analytical Separations
Analytical separations may ‘blur the line’ between analysis
and purification
You can safely say your separation is analytical when the it
tells you something specific about your analyte
Examples of ‘sometimes’ analytical separations include:
Chromatographic:
Thin Layer (TLC) / Paper
Size Exclusion (SEC) / Affinity
Electrophoretic:
Agarose/Polyacrylamide Gel
Capillary Electrophoresis (CE)
Analytical ultracentrifugation
The Beginning: Paper Chromatography
The unifying aspect of chromatographic methods is that the
separation is not with an applied electric field
The very first type of chromatography was the ‘adsorption’
variety, which includes paper and TLC
Separated chlorophylls
and karatenoids using
calcium carbonate as the
Mikhail Tsvet ‘stationary phase’
1872-1919
About 20 years later, Richard L.M.
Synge and Archer Martin develop
paper chromatography and win the
nobel prize
Adsorption Chromatography Mechanism
Adsorption chromatography uses a stationary phase and a
mobile phase
The stationary phase is usually a solid ‘sheet’ either cellulose
(paper), silica gel (standard TLC) or aluminum oxide
The stationary phase must have ‘moderately charged’
functional groups:
N
Cellulose
Silica Gel
Al2O3
C
Adsorption Chromatography Mechanism
To move the analytes over the stationary phase, we use a
non-polar solvent which is drawn up via capillary action
Some popular mobile phase solvents are:
Non-polar: Alkanes (up to octane), Diethyl ether
Moderate polarity: Alcohols, Ketones (not acetone)
Polar: H2O, Weak Salt Buffers
TLC and Paper Visualization
Once you’ve run the molecule, how do you see it?
If you’re very lucky, then your analytes are chromophoric
If you’re somewhat lucky, then your analytes absorb UV light
If you’re unlucky then you have to use a stain (like iodine)
TLC and Paper Chromatography Nowadays
In Biochemistry, TLC and paper chromatography had their
heyday in the 50s (Remember Sanger).
Nonetheless they are still routinely used as basic analytical
tools, particularly in organic chemistry (TLC)
We don’t separate proteins this way – electrophoretic
separations work much better
“Paper Chromatography” 2000-2007: 175 results
1960-1967: 720 results
“TLC” 2000-2007: 420 results
1980-1987: 394 results
Column Chromatography: Size Exclusion
Adsorption chromatography can also be done on a column,
but more for separation than analysis
Probably the most common chromatography technique for
proteins is ‘size exclusion’
It is also called ‘gel
permeation’ chromatography
Column is usually agarose
beads (i.e. Sephadex™)
Size Exclusion Examples
Analytical Size exclusion is usually used to distinguish the
oligomeric state of proteins
Chromatogram
C-CIC-3
Standards
Mol. Wt.
Biochemistry (2007), 46 (51): 14996-15008
C-CIC-3 is about 20.5 kDa but the
analysis shows 23kDa. Loose structure?
Standards Stokes
Radius
J. Biochem. (2006), 139 (5): 813-820
Affinity Chromatography
Affinity chromatography comes in a few flavors: Immuno-,
Immobilized Metal Ion Affinity (IMAC) and just plain
affinity (e.g. GST)
The difference between this and adsorption is that here the
analyte actually sticks to the column until it is washed off
Cu2+ / His6 and GST /
Glutathione are very
common ways of
purifying proteins
Nonetheless, there are
some examples of
analytical affinity
chromatography
Affinity Chromatography Examples
Uses the affinity of glucose for boronate to detect the extent
of glucosylation of Ig2 antibodies
Anal. Chem. (2007) 79 (24): 9403-9413
TAP Tagging
Tandem Affinity Purification (TAP) is a powerful purification
technique that analyzes protein/protein interactions
Methods (2001) 24, 218–229
Chromatography Instrumentation
The Analytical Chromatography instrument is the FPLC
The Agilent 1200 System
The GE ÄKTA
Electrophoresis Theory
Electrophoresis uses an electric current or field to ‘push’
analytes through a medium
In the simplest case, the electrophoretic mobility of an
analyte e is proportional to the electric field E.
v
e
E
In reality, though, electrophoretic mobility has to take into
account the ‘double layer’ - solvent ions of oposite charge
that cluster around the analyte.
Electrophoresis Theory
The effect of the double layer for a given solvent and analyte
are incorporated into the zeta potential term .
Where is the dielectric constant of the
solvent, 0 is the permitivity of the vaccum
(8.85*10-12) and is the dynamic vicocity
0
e
So far we have assumed that we are dealing with particles
whose size is on the order of the double layer. But our
analytes are much bigger. In this case, we are more
concerned with the stokes radius relative to the amount of
charge.
z
Where z is the charge, is the
ue
6r viscosity and r is the Stokes radius
Electrophoresis of Nucleic Acids
If we’re trying to separate nucleic acids, we have a problem
because:
They are always negatively charged at neutral pH.
All molecules will travel in the same direction
Each additional nucleoside confers an additional charge,
so charge is directly proportional to size.
All molecules will have the same e
The solution is to use a gel which consists of pores surrounded
by cross-linked fibers
This will make e dependent
on the Stokes radius
EM image of
Agarose Gel
DNA/RNA Electrophoresis
Double stranded DNA or RNA are molecules that repel
themselves. They will all form rod-like structures.
So now we can separate our nucleic acids on the basis of
size. We can visualize with a fluorescent dye (usually
ethidium bromide) and compare to a standard to get a
relatively good quantitative size:
Electropherogram
Single stranded DNA or RNA may interact with itself
forming a ‘supercoil’. e for supercoiled DNA or RNA is a
mite unpredictable.
Protein Electrophoresis
Proteins are even trickier than DNA/RNA:
They are all effectively supercoiled (3° Structure)
They can be either positively or negatively charged
The number of charges depends on the amino acid sequence
and is not proportional to size.
The solution is to expose
the protein so a detergent
(usually sodium dodecyl
sulphate - SDS)
-ve
+ve
Protein Electrophoresis
Proteins will bind an amount of SDS roughly proportional to
their size (1.4g/g polypeptide), so we have effectively
created the same situation as for DNA/RNA electrophoresis.
The
‘Standard’
ladder
The OverExpressed
protein of
interest?
The ‘gel of choice’ for SDS-page
protein electrophoresis is
polyacrylamide. Danger!
Acrylamide monomers are
potent neurotoxins
Sometimes proteins are preboiled in a reducing agent to
eliminate S-S.
Proteins are visualized using a dye (coomassie blue)
or other stain (silver)
2D Electrophoresis
If you want to analyze the whole protein content of a cell, a
1D separation just isn’t gonna do it.
The solution is to do a 2D separation using a gel
that has a pH gradient.
Proteins will run on this gel (in both directions)
until they hit their isoelectric point where they
aggregate
Then you can soak the gel in
SDS, flip the voltage 90° and
-ve
separate by size.
+ve
2D Gels
2D Electrophoresis is a powerful tool for analyzing the
protein complement of simple cells
Capillary Electrophoresis
You can also make molecules pass through a narrow (usually
fused silica) capillary
Typically, your analyte is in a buffer with net +ve charge,
which generates an electroosmotic flow (EOF) towards the
cathode
EOF overpowers e, thus all analytes move towards the
cathode at rates dependent on their e
Capillary Electrophoresis
The advantages of CE are:
Really good separation of slow diffusing analytes
Rsep
eV
1 e D
4 (e o )
Where: e is the difference in
apparent electrophoretic mobility,
e is the average electrophoretic
mobility, V is the applied voltage
and D is the diffusion coefficient
Resolution is limited by Diffusion coefficient and flow
rate
Very, very low sample consumption
Analytical Ultracentrifugation
In analytical ultracentrifugation, analytes are separated on
the basis of their sedimentation when they experience a
centripetal force.
Usually carried out at speeds around
60,000
Analytical Ultracentrifugation
Or, described mathematically…
angular velocity radius from center of spin
mass of a single molecule
Fs m 2 r
M 2
r
N
This is counteracted by the Boyant force (due to displacement
of solute) and the frictional force
sedimentation velocity
coefficient of friction
mass of solute displaced
Fb m0 2 r
M
m0
N
Solvent
density in
g/mL
Volume occupied by 1g of solute
F f f s
M (1 ) s
2 s
Nf
r
Sedimentation
coefficient
Sedimentation Velocity Experiments
The most basic type of ultracentrifugation experiment is to
measure the rate at which the analyte moves away from the
center of rotation
What is actually measured is the movement
of the boundary between dissolved analyte
and ‘empty’ buffer c 1 c
M (1 ) s
2 s
Nf
r
f
Mr
RT
ND
RTs
D (1 )
t
2 2
rD
s
r c
r r r
Sedimentation Equilibrium
In sedimentation equilibrium, an equilibrium is established
between sedimentation away from the center of rotation
and diffusion towards the center of rotation.
C A ( r ) C A , 0 e ( r
M (1 ) 2
RT
2
r02 ) / 2
Density Gradient Centrifugation
Density gradient is similar to equilibrium sedimentation
except there is a permanent solvent density gradient. This
sharpens the contrast between the sedimentation equilibria
of samples with the same shape, but slightly different
masses:
Meselson and Stahl, P.N.A.S., 44,
(1958), 671-682)
 0
0