2013.12.19 Local anaesthetic
advertisement

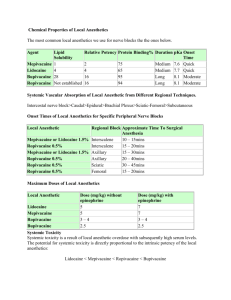
LOCAL ANAESTHETIC AGENT By Dr Nur Hafizah binti Ibrahim Moderator: Dr Najwa binti Mansor OUTLINE OF PRESENTATION • • • • • • • • Definition History Commercial Preparations Structure-activity Relationships Mechanism of Action Minimum Concentration Pharmacokinetics Toxicity DEFINITION • A drug that produce reversible conduction blockade of impulses along central and peripheral nerve pathways after regional anesthesia, producing autonomic nervous system blockade, sensory anesthesia and skeletal muscle paralysis in the area innervated. • Interruption of transmission of autonomic, somatic sensory and somatic motor impulses. • Removal of the local anesthetic is followed by spontaneous and complete return of nerve conduction, with no evidence of structural damage to nerve fibers as a result of the drug’s effects. HISTORY • Cocaine – 1st local anesthetic introduced by Kollar in 1884, for use in opthalmology. – It is a naturally occurring compound extracted from leaves of Erythroxylon coca, a plant growing in the Andes Mountains. – Halsted recognized the ability of injected cocaine to interrupt nerve impulse conduction, leading to the introduction of peripheral nerve block anesthesia and spinal anesthesia. – Another unique feature of this drug is the ability to produced localized vasoconstriction. • 1st synthetic local anesthetic was the ester derivative procaine, introduced by Einhorn in 1905. • Lidocaine was synthesized as an amide local anesthetic by Lofgren in 1943. – It produces more rapid, intense and longer-lasting effect that procaine. – It is also effective topically and a highly efficacious cardiac antidysarhythmic drug. COMMERCIAL PREPARATIONS • Poorly soluble in water – marketed most often as water-soluble hydrochloride salt. • These HCl salts are acidic (pH 6) – contributing to the stability of LA. • An acidic pH also important if epinephrine is present in LA solution, because this cathecolamine is unstable at alkaline pH. • Sodium bisulphite (strongly acidic), may be added to LA-ephephrine solutions to prevent oxidative decomposition of epinephrine. STRUCTURE-ACTIVITY RELATIONSHIPS • Consist of a lipophilic and a hydrophilic portion separated by connection hydrocarbon chain. • Lipophilic group: – usually unsaturated aromatic ring such as paraaminobenzoic acid. – Essential for anesthetic activity • Hydrophilic group: – Usually a tertiary amine (diethylamine). • A hydrocarbon chain (consist of ester or amide) links both portion to produce delicate balance between lipid solubility and water solubility Basic structure Classifications of LA ESTER • Procaine • Tetracaine • Amethocaine • Cocaine AMIDE • Lidocaine • Prilocaine • Bupivacaine • Articaine • Mepivacaine ***If the local anesthetic has two ‘i’s in its name, it’s an amide Properties of LA Agents PROPERTIES ESTERS AMIDES Metabolism Rapid – plasma cholinesterase Less likely Slow – hepatic Allergic reaction Possible – PABA derivatives Very rare Stability in solution Breaks down in ampules (heat, sun) Very stable chemically Onset of action Slow as a general rule Moderate to fast pKa’s Higher than pH = 7.4 (8.5-8.9) Close to pH = 7.4 (7.6-8.1) Systemic toxicity More likely Physicochemical properties • The chemical structure and physicochemical characteristics of LA affect their clinical properties. • Modification of the chemical structure (lengthening of the hydrocarbon chain within critical length or increasing the number of carbon atoms in the aromatic ring or tertiary amine) may alter lipid solubility, potency, rate of metabolism & duration of action • In particular, these are modified by: – Lipid solubility – Protein binding – Dissociation constant (pKa value) • Lipid solubility – Lipid solubility of diff anesthetics governs their ability to penetrate perineuronal tissues and neural membrane, and reaches their site of action in neuroplasm – More lipid soluble penetrates membrane more easily, less molecule requires for nerve conduction blockade i.e. more potent – E.g. Bupivacaine, levobupivacaine and ropivacaine are app 3-4x as potent as lidocaine or prilocaine due to differences in their lipid-solubility • Protein binding: – Tissue protein binding primarily affect the duration of action of LA • plasma protein binding > longer duration of action – Plasma protein binding acts as depot – Tissue receptor binding – removed at a slower rate – Examples: • Procaine is not extensively bound to tissue protein, has short duration of action • Bupivacaine, levobupivacaine & ropivacaine are extensively bound to plasma and tissue protein --- prolonged effect • Dissociation constant (pKa value) – pKa is equal to pH at which the concentration of ionized base and non-ionized base are equal. – It can be used to calculate the proportion of 2 forms that are present in solution at diff pH values pH = pKa + log 10 (BH+) / (B) – For weak base, (unionised ) / (ionised) = 10 (pH- pKa) • • • Is the most important factor affecting rapidity of onset of action. pKa value governs the proportions of LA that is present in non-ionized form at physiological pH values and therefore available to diffuse across tissue barrier to its site of action. LA with a pKa near physiological pH will have a greater degree of unionized molecules → More LA diffused across membrane → rapid onset of action MECHANISM OF ACTION • Modulated-receptor theory • Frequency-dependent blockade • Guarded-receptor theory • Membrane Expansion theory • Surface-charge theory Modulated-receptor Theory • Bindings of LA is stereospecific and dependent on the conformation state of the Na channel. • 3 functional states: – Activated, open – Inactivated/refractory, closed – Resting, closed • LA has high affinity for the open and inactivated closed state and low affinity for the rested closed state. • LA prevents transmission of nerve impulses (conduction blockade) by inhibiting passage of Na ions through ion-selective Na channels in nerve membrane. • Na channel itself is a specific receptor for LA molecules. • So, occlusion of open Na channel by LA molecules contributes little to overall inhibition of Na permeability. • Failure of Na ion channel permeability to increase will slows the rate of depolarization such that threshold potential is not reached and thus an action potential is not propagated Frequency-dependent blockade • Defines a situation where the more frequent the channel are activated, the greater the degree of block produced. • During action potential, LA exhibit higher affinity to receptor at Na channel. • But when action potential is over, affinity is less and some unbind. • If another action potential (frequency) arrives before all drugs unbound, and additional increment of blocking will occur. Guarded-receptor Theory • The affinity towards receptor is constantly high in contrast to first theory which depend on conformational state of Na channel. • However the access of LA to the receptor is limited by channel. Membrane expansion theory • Lipophilic LA incorporated into lipid bilayer causing a volume expansion & distortion to the conformation of axonal membrane and hence the Na channel resulting in its inactivation. • This is mediated by unionized forms. Surface charge theory • The protonated form of LA neutralized the negative charge of membrane of the nerve which resulting in hyperpolarizing the nerve. MINIMUM CONCENTRATION • Minimum concentration (Cm) of LA necessary to produce conduction blockade of nerve impulses. • It is analogous to the MAC for inhaled anesthetic. • Influenced by: – – – – nerve diameter (↑D ↑Cm), Myelinated nerve ↑Cm Frequency of nerve stimulation (↑F ↓Cm) Tissue pH (↑pH ↓Cm) • Differential Conduction Blockade: – Is illustrated by selective blockade of preganglionic sympathetic nervous system B fibers using low concentration of LA. – Slightly higher concentration of LA interrupt conduction in small C fibers and small-and-medium sized A fibers, with loss of sensation for pain and temperature. – However touch, proprioception and motor function still intact. – Sometimes misinterpreted as failure of LA in an anxious patient. PHARMACOKINETICS • • • • Absorption Distribution Metabolism Excretion Absorption • Influenced by : – Site of administration – Dosage – Vasoconstrictor effect – Pharmacologic characteristic of the drugs – Patient’s factor age, cardiovascular status and hepatic function. • Site of administration: – Intravenous – Mucuos membrane/trachea – Intercostal block – Caudal block – Epidural block – Brachial plexus block – Peripheral nerve block – Subcutaneous infiltration fastest slowest • Dosage – Blood level of LA is related to total dose of drug rather than specific volume or conc of solution – Linear relationship between total dose & peak blood concentration achieved • Patient’s factor – In pregnancy ↑ LA effect d/t ↓ protein binding – In abscess ↓ LA effect d/t more ionized fraction in acidic pH – In hyperkalemic state ↑ LA effect d/t more inactive channel • Vasoconstrictor effect: – Vasoconstriction at site of injection – ↓ rate of absorption, ↓ Systemic toxicity, ↑ duration of action – By addition of adrenaline 5μg/ml (1:200 000) – Higher Dosage offers no additional benefits but increases symphatomimetic activites – Ropivacaine & Cocaine has intrinsic vasoconstrictor activities – Lignocaine, mepivacaine, bupivacaine, etidocaine exhibit vasodilator effects • all local anesthetics possess some degree of vasoactivity; most producing some level of vasodilation • ester local anesthetics are potent vasodilating drugs • Procaine (Novocaine) possesses tremendous vasodilating abilities which are employed to halt arteriospasm (accidental IA injection) Specific properties of the drugs ↑ Lipid solubility reduce rate of absortion Degree of ionization reduced protein binding ↓ pKa ↓ molecular weight Distribution • Depends on organ uptake, which determined by: – Tissue perfusion – Tissue/blood partition coefficient – Tissue mass – Lung extraction – Placental transfer • Tissue perfussion: – highly diffuse organ (brain, lungs, liver, kidney & heart) are responsible for initial rapid uptake – Followed by slower redistribution to moderately perfused tissue (muscle & gut) • Tissue/blood partition coefficient – strong plasma protein binding eg Bupivacaine, tends to retain LA in the blood – Influence by changes in concentration of alpha1 acid glycoprotein (eg; pregnancy, old age, concurrent liver disease) – high lipid solubility; facilitates tissue uptake • Tissue mass: – muscle provides greatest reservoir for LA agents d/t its large mass • Lung extraction: – First pass pulmonary extraction esp lidocaine, bupivacaine & prilocaine. – Limit the concentration of drug that reaches systemic circulation for distribution to the coronary and cerebral circulation. • Placental transfer: – Highly plasma protein binding LA limits diffusion across placenta – Esters undergo rapid hydrolysis hence not available for transfer across placenta – Acidosis in fetus, which may occur during prolonged labour, can result in accumulation of LA molecules in the fetus (ion trapping) – This results in an accumulated concentration of drug in the fetus for two reasons. • once the drug becomes ionized it cannot readily diffuse back across the placenta. This is known as ion trapping. • a concentration gradient of nonionized drug is maintained between the mother and the fetus. Ion trapping • Fetal blood is slightly more acidic than maternal blood, with a pH about 0.1 unit less than maternal blood pH. • The lower pH of fetal blood facilitates the fetal uptake of drugs that are basic. • Weakly basic drugs, such as local anesthetics and opioids, that cross the placenta in the nonionized state become ionized in the fetal circulation Metabolism Metabolism of ESTER • Undergo hydrolysis by cholinesterase enzyme, in the plasma and liver • Rate of hydrolysis varies, and resulting metabolites are pharmacologically inactive • PABA may be associated with allergic reaction • Systemic toxicity is inversely propotional to the rate of hydrolysis • In SAB, in view of lack of cholinesterase enzyme in cerebrospinal fluid, so the termination of action of intrathecal LA depends on absorption into the blood stream. • Prolonged in neonates, liver dysfunction, ↑BUN, parturient and atypical plasma cholinesterase homozygotes activity only ~50% in the newborn and doesn’t reach adult values until about 1 yr Metabolism of AMIDE • Metabolized by microsomal enzyme located primarily in the liver. amide base aminocarboxilic acid + cyclic aniline derivative ↓ ↓ N-dealkylation hydroxylation • The rate of metabolism are more slower and complex as compared to ester • Systemic toxicity are more likely. • initial reaction N-alkylation subsequently hydrolysis – exception to Prilocaine – hydrolysis take place first forming o-toluidine – further metabolized to 4 and 6-hydroxytoluidine – o-toluidine responsible in metHb in high doses • Benzocaine also can cause metHb • Rx: methylene blue ( Fe3+ Fe2+ ) COCAINE • an alkaloid derived from leaves of Erythroxylon coca • at low dose : – produces a feeling of well-being and euphoria – BRADYCARDIA d/t central vagal stimulation – block uptake of noradrenaline results vasoconstriction and mydriasis • At higher dose: – stimulates vomiting center – central and peripheral sympath stimulation • tachycardia • ↑ capacity muscular work – eventually causes • • • • • • Convulsion coma myocardial depression medullary depression respiratory depression VF and DEATH • cocaine blocks conduction when applied directly to nerve tissue & may therefore be used in surface anesthesia • cocaine was used in the past for ophthalmological procedures, however has now been abandoned • due to sloughing of the corneal epithelium & increased intraocular pressure • the only use today is as a topical local anaesthetic in ENT (5%) • cocaine itself constricts blood vessels and the use of adrenaline is contraindicated as it sensitizes the myocardium Chloroprocaine • Chloroprocaine is halogenated Procaine with additional chlorine atom • short duration of action d/t ↑ 3-4 x rate of metabolism • low toxicity • less allergenic compare to procaine • rapid onset • is a halogenated derivative of procaine and has similar pharmacological properties • the rapid plasma hydrolysis results in a short duration of action and low toxicity (potency = 1.0) • onset of action is ~ 6-12 mins and the duration ~ 60 mins, depending upon the administered dose • maximal recommended doses are 1000 & 800 mg, with and without adrenaline • it is ineffective for topical anaesthesia but suitable for infiltration, or peripheral nerve block in a 1.0% solution • the low toxicity, short duration, rapid metabolism and low foetal:maternal blood partitioning make it a suitable choice in obstetrics • suspected neural toxicity with epidural/spinal administration actually related to the addition of sodium bisulphite to the adrenaline containing solutions • this was enhanced by the low pH of the solution and the limited buffering in the subarachnoid space Tetracaine • this is also an ester of PABA but is a potent, long acting agent which is hydrolysed at a much slower rate • its actions are similar to those in this group • suitable for spinal anaesthesia, hypo & hyperbaric, in a 1.0% solution when the dose ranges from 5.0 to 20 mg • onset of action is ~ 10 mins and the duration of action 60 to 90 mins • maximum recommended dose is ~ 100 mg for a 70 kg male • NB: this is a useful agent when the amide agents are contraindicated Lignocaine • Etidocaine derived from Lignocaine subtitution of ethyl group make it 50x more lipid soluble ↑2-3x duration of action (more longer than Bupi) • Structure – Amide LA; derivative of diethylaminoacetic acid • Presentation – Clear aq solution lidocaine hydrochloride – Plain 0.5% (local infiltration) 1%, 2%(nerve blocks, extradural anaesthesia) – with adrenaline 1:200 000 – Gel 2% with or without chlorhexidine – 4% aq solution for topical application to pharynx, larynx, trachea – 10% spray for oral cavity & upper resp tract • Recommended max dose 3mg/kg (7mg/kg with adrenaline) • Acute ventricular dysrhythmia (class 1) – 1mg/kg over 2 min followed by infusion 4mg/min (1st hr), 2mg/min (2nd hr) & subsequently 1mg/min. • Reduce stress response – 1-2mg/kg IV 5min before intubation • Clinically: – acts in 2-20min; duration 60-120min depending on conc, vasoconst – CVS therapeutic & toxic effect – RS bronchodilation in subtoxic dose • Pharmacokinetic MW pKa Part coef 234 7.9 2.9 • Absorption – bioavailability by oral route is 24-46% due to high extraction & 1st pass hepatic metabolism • Distribution Prot bind Vd 70% 0.75-1.5L/kg • Metabolism – 70% metabolised in liver by Ndealkylation to MEGX, GX with further hydrolysis prior to renal excretion • Excretion - <10% excreted unchanged • Cl – 6.8-11.6ml/kg/min t½ - 90-110min Bupivacaine has butyl (C4H9) group to the piperidine N2, makes it more potent 35 times and longer action 3-4 times compare to Mepivacaine (methyl – CH3) • Structure – amide LA, pipecoloxylidide group, racemic • Presentation – clear colourless, aq solution (bupivacaine hydrochloride ) – Plain (0.25%, 0.5%, 0.75%) – With 1:200 000 (5µg/ml ) adrenaline – Heavy 0.5% with 80mg/ml dextrose (SG 1.026) • Recommended max dose 2mg/kg (150mg plus up to 50mg 2 hourly subsequently) • Clinical - acts within 10-20min and almost immediate with intrathecal admin, duration of action 4-8hrs. 4X as potent as lignocaine • • • • • • Pharmacokinetics MW pKa Part coef 288 8.1 27.5 Absorption addition of adrenaline does not influence rate of systemic absorption Distribution : Prot bind Vd 95% 1L/kg(41-103L) Metabolism liver microsomal enzymes P450 to 2,6Pipecoloxylidine (N-deakylation), N-desbutyl bupivacaine & 4OH bupivacaine also formed Excretion 5% excreted as PPX, 16% excreted unchanged in urine Cl : 0.47L/m t½ : 0.31-0.61Hr (after IV admin) • postoperative analgesia can be maintained for 3-4 hours with epidural blockade • peak plasma levels following caudal or epidural administration are reached within 30-45 minutes, followed by decline over 3-6 hours • although more toxic, a lower foetal/maternal ratio has been observed cf. other agents • thus, bupivacaine is the recommended for obstetric analgesia/anaesthesia, with the exception of paracervical block • however, caution should be used with repeated doses • similarly, renal disease is unlikely to affect bupivacaine clearance in the short term (~ 24 hrs), • however, toxicity may result with prolonged or repeated administration Levobupivacaine • • • • Levobupivacaine is S enantiomer of Bupivacaine Less cardio and CNS toxicity Less vasodilation 13% Less potent than bupivacaine • less prolonged motor blockade but longer sensory blockade after epidural administration Ropivacaine ropivacaine is single-enantiomer structurally resemble of Bupi/Mepivacaine with propyl (C3H7) group to the piperidine N2 atom • Ropivacaine is pure S-enantiomer of Bupivacaine • It’s potency, onset time and duration of action almost identical to bupivacaine • less motor block • larger therapeutic index • 70% less cardiotoxic contribute fr its lower LIPID SOLUBLE and its availability as S-isomer. • Structure – Amide LA, pipecoloxylidide group, pure S-enantiomer • Presentation – Clear colourless solution containing 0.2/ 0.75/ 1.0% ropivacaine hydrochloride • Recommended dose – 3.5mg/kg, 250mg (150mg for C-section under epidural), Cummulative dose 675mg over 24h according to data so far; not currently intended for intrathecal admin and in children < 12 years • Clinical – sensory blockade similar in time course to that produced by bupivacaine; motor blockade is slower in onset & shorter in duration than after an equivalent dose of bupivacaine; less cardiotoxic than bupivacaine; Intrinsic vasoconstrictor, mild CNS Sx • Pharmacokinetics MW 274 • Distribution : Prot bind 94% pKa 8.1 Part coef 6.1 Vd 0.8L/kg (52-66L) • Metabolism occurs in liver to 2,6 pipecoloxylidine (Ndeakylation), Aromatic hydroxylation to 3 & 4OH ropivacaine • Excretion 86% (mostly conjugated) excreted in the urine, 1% unchanged Cl – 0.82L/m t½ - 59-172min Prilocaine • Prilocaine (CITANEST) • equipotent to lignocaine with a longer duration of action and lower toxicity • rapid plasma hydrolysis makes it the agent of choice for regional IV blockade • causes methaemoglobinaemia in high doses but this is rarely clinically significant • used in all types of anaesthesia in similar concentrations to lignocaine Mepivacaine • used for infiltration, nerve block, caudal & epidural anaesthesia but is ineffective topically • similar potency and onset to lignocaine but slightly longer duration of action • used in similar concentrations to lignocaine • not recommended for obstetric use due to high foetal:maternal ratio Etidocaine • is a long acting derivative of lignocaine, with much higher lipid solubility and protein binding • it is ~ 2-3 times more potent than lignocaine but less toxic than bupivacaine • effective in infiltration, nerve block, epidural & caudal anaesthesia • results in a high degree of motor blockade when administered into the epidural space EMLA • eutectic mixture of local anaesthetics is a 5% mixture of lignocaine (25 mg/g) &prilocaine base (25 mg/g) • CONTACT TIME AT LEAST 1 HOUR • Duration of action 1-2 hr • Typically 1-2g applied to 10cm² area of skin maximum 2000cm² in adult and 100cm² for child <10kg TOXICITY • Occur due to excessive plasma and tissue concentrations of the LA. • Common cause include accidental intravascular injection and overdose. The relationship between lignocaine and plasma concentration and pharmacological effects Signs of toxicity BEGIN with numbness of the tongue/circumoral tissue restless,vertigo,tinnitus,dizziness slurred speech, skeletal ms twitching (face & extrimities) seizure-tonic-clonic drowsiness hypotension & apnea ASYSTOLE CNS toxicity • A relatively small dose (5 to 10ml lignocaine 1%) may cause ringing in the ears, a metallic taste in the mouth, numbness of the tongue, blurring of vision, drowsiness with decreased awareness, muscular twitching, restlessness and apprehension. • With a larger dose, delirium may ensue and progress to grand mal convulsions. • Asphyxia may develop if the convulsions are not treated rapidly and adequately. Cardiovascular toxicity • In general LA depress myocardial automaticity • LA also ↓ duration of refractory period • At higher dose LA depress contractility and conduction velocity • Prolongation PR and QRS interval on ECG • Myocardial depressant and vasodilatation lead to HYPOTENSION • Cardiac dysrhythmia or circulatory collapse is often the presenting sign of LA overdose during GA • Lignocaine is a less potent LA as compared to bupivacaine or ropivacaine and a higher dose is required to cause cardiovascular and CNS toxicity. The ratio of the dosage required for irreversible cardiovascular collapse (CVS) and the dosage that will produce CNS toxicity (convulsions), i.e. the CVS/CNS ratio, is lower for bupivacaine than for lignocaine. • CVS/CNS dose ratio: – lignocaine 7.1 – bupivacaine 3.7 • This indicates that 7 times as much lignocaine is required to induce irreversible CVS collapse as is needed to produce convulsions Treatment of LA toxicity • Depends on severity, minor reaction - resolve spontaneously • If seizures – protect airways, abort the seizures by midazolam, thiopentone, propofol • If cardiac arrest - follow ACLS • IF NOT RESPONSIVE, intravenous lipid or cardiopulonary bypass may be considered Management of severe LA toxicity by the Australian and New Zealand College of Anaesthetists (AN ZCA ) Intravenous 20% lipid emulsion • Was invented by Dr Weinberg, 1998 • Rosenblatt 2006, first published rescue • Officially promoted as a treatment in Irelend and British • The mechanism is not clear yet • it serve as lipid sink which capture the LA molecules making then unavailable to tissue • Changes during pregnancy: – Increased sensitivity (more rapid onset of conduction blockade) may be present during pregnancy. – Alteration in protein-binding characteristics of bupivacaine may result in increased concentration of pharmacologically active unbound drug in the parturient’s plasma. – Nevertheless, progesterone, which binds to the same alpha1-acid glycoprotein as bupivacaine, does not influenced protein binding of this LA. – This evidence suggest that bupivacaine and progesterone bind to discrete but separate sites on protein molecules.