Part III
advertisement

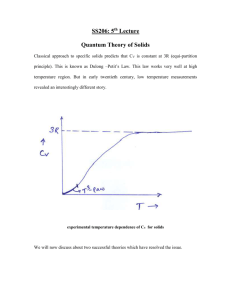
Quantum Mechanical Simple Harmonic Oscillator • Quantum mechanical results for a simple harmonic oscillator with classical frequency ω: The energy is quantized 1 E nn n • Energy levels are equally spaced! E 2 n = 0,1,2,3,.. Often, we consider En as being constructed by adding n excitation quanta of energy to the ground state. 1 E 0 2 Ground state energy of the oscillator. If the system makes a transition from a lower energy level to a higher energy level, it is always true that the change in energy is an integer multiple of Phonon absorption or emission ΔE = (n – n΄) n & n ΄ = integers In complicated processes, such as phonons interacting with electrons or photons, it is known that phonons are not conserved. That is, they can be created and destroyed during such interactions. Thermal Energy & Lattice Vibrations As we’ve been discussing in detail, the atoms in a crystal vibrate about their equilibrium positions. This motion produces vibrational waves. The amplitude of this vibrational motion increases as the temperature increases. In a solid, the energy associated with these vibrations is called Thermal Energy • A knowledge of the thermal energy is fundamental to obtaining an understanding many of the basic properties of solids. A relevant question is how do we calculate this thermal energy? • Also, we would like to know how much thermal energy is available to scatter a conduction electron in a metal or semiconductor. This is important; this scattering contributes to electrical resistance in the material. • Most important, though, this thermal energy plays a fundamental role in determining the Thermal Properties of a Solid • A knowledge of how the thermal energy changes with temperature gives an understanding of the heat energy which is necessary to raise the temperature of the material. • An important, measureable property of a solid is it’s Specific Heat or Heat Capacity Lattice Vibrational Contribution to the Heat Capacity The thermal energy is the dominant contribution to the heat capacity in most solids. In non-magnetic insulators, it is the only contribution. Some other contributions: Conduction Electrons in metals & semiconductors. The magnetic ordering in magnetic materials. Calculation of the vibrational contribution to the thermal energy & heat capacity of a solid has 2 parts: 1. Evaluation of the contribution of a single vibrational mode. 2. Summation over the frequency distribution of the modes. Vibrational Specific Heat of Solids cp Data at T = 298 K Thermal Energy & Heat Capacity Einstein Model _ Pn n Average energy of a harmonic oscillator and hence of a lattice mode of angular frequency at temperature T n Energy of oscillator 1 n n 2 The probability of the oscillator being in this level as given by the Boltzman factor exp( n / kBT ) _ Pn n n 1 1 n exp n / k BT _ 2 2 n 0 1 exp n / k BT 2 n 0 1 z exp[( n ) ] 2 k BT n 0 z e / 2 k BT e 3 / 2 k BT e 5 / 2 k BT z e / 2 k BT (1 e / k BT e 2 / k BT z e / 2 k BT (1 e / k BT 1 ..... ..... ) According to the Binomial expansion for x«1 where x / kBT (*) Eqn (*) can be written 1 z k BT 2 (ln z ) z T T _ e / 2 k BT 2 k BT ln T 1 e / kBT _ k BT 2 / 2 k BT / k BT ln e ln 1 e T _ / k BT k BT 2 ln 1 e T 2 k T T B k B / k BT 2k k 2T 2 e 1 _ e 2 B B k BT 2 2 / k BT 4 k T 2 B 1 e 1 e _ k BT 2 _ Finally, the result is 1 2 e / k BT / k BT / k BT 1 x' (ln x) x x _ 1 2 e / k BT 1 This is the Mean Phonon Energy. The first term in the above equation is the zero-point energy. As mentioned before even at 0ºK atoms vibrate in the crystal and have zero-point energy. This is the minimum energy of the system. The average number of phonons is given by the Bose-Einstein distribution as _ (number of phonons) x (energy of phonon) = (second term in ) 1 n( ) e kBT 1 The second term in the mean energy is the phonon contribution to the thermal energy. kBT Mean energy of a harmonic oscillator as a function of T 1 2 T Low Temperature Limit kBT 1 2 e kBT 1 _ _ 1 2 Since exponential term gets bigger Zero Point Energy kBT Mean energy of a harmonic oscillator as a function of T 1 2 x2 e 1 x .......... 2! x T e 1 High Temperature Limit k BT _ 1 << kBT 2 is independent of frequency of oscillation. 1 1 k BT This is the classical limit because the energy _ 1 steps are now small compared with the energy k BT 2 of the harmonic oscillator. k BT So that is the thermal energy of the classical 1D harmonic oscillator. _ kBT Heat Capacity C • Heat capacity C can be found by differentiating the average phonon energy _ 1 2 e 1 kBT d Cv dT Let kB k BT e kBT e 2 1 k 2 kBT Cv k B 2 k T B 2 e kBT e eT Cv k B T e T 1 2 2 k BT 1 2 eT Cv k B T e T 1 2 2 where k Specific heat in this approximation vanishes exponentially at low T and tends to classical value at high temperatures. Cv kB 2 Area = kB T These features are common to all quantum systems; the energy tends to the zero-point-energy at low T and to the classical value of Boltzmann constant at high T. Specific heat at constant volume depends on temperature as shown in figure below. At high temperatures the value of Cv is close to 3R, where R is the universal gas constant. Since R is approximately 2 cal/K-mole, at high temperatures Cv is app. 6 cal/K-mole. 3R Cv T, K This range usually includes RT. From the figure it is seen that Cv is equal to 3R at high temperatures regardless of the substance. This fact is known as Dulong-Petit law. This law states that specific heat of a given number of atoms of any solid is independent of temperature and is the same for all materials! Cv vs T for Diamond Points: Experiment Curve: Einstein Model Prediction Classical Theory of Heat Capacity of Solids The solid is one in which each atom is bound to its side by a harmonic force. When the solid is heated, the atoms vibrate around their sites like a set of harmonic oscillators. The average energy for a 1D oscillator is kT. Therefore, the averaga energy per atom, regarded as a 3D oscillator, is 3kT, and consequently the energy per mole is = 3NkBT 3RT where N is Avagadro’s number, kB is Boltzmann constant and R is the gas constant. The differentiation wrt temperature gives; d Cv dT Cv 24.9 Cv 3R 3 6.02 1023 (atoms / mole) 1.38 1023 ( J / K ) J ;1J 0.2388Cal Cv ( K mole) 6 Cal ( K mole) Einstein heat capacity of solids • The theory explained by Einstein is the first quantum theory of solids. He made the simplifying assumption that all 3N vibrational modes of a 3D solid of N atoms had the same frequency, so that the whole solid had 2 a heat capacity 3N times e Cv k B T T e T 1 2 • In this model, the atoms are treated as independent oscillators, but the energy of the oscillators are taken quantum mechanically as This refers to an isolated oscillator, but the atomic oscillators in a solid are not isolated.They are continually exchanging their energy with their surrounding atoms. • Even this crude model gave the correct limit at high temperatures, a heat capacity of the Dulong-Petit law where R is universal gas constant. 3NkB 3R • At high temperatures, all crystalline solids have a specific heat of 6 cal/K per mole; they require 6 calories per mole to raise their temperature 1 K. •This arrangement between observation and classical theory break down if the temperature is not high. •Observations show that at room temperatures and below the specific heat of crystalline solids is not a universal constant. Cv 6 cal Kmol kB Cv 3R In each of these materials (Pb,Al, Si,and Diamond) specific heat approaches constant value asymptotically at high T. But at low T’s, the specific heat decreases towards zero which is in a complete contradiction with T the above classical result. • Einstein model also gave correctly a specific heat tending to zero at absolute zero, but the temperature dependence near T= 0 did not agree with experiment. • Taking into account the actual distribution of vibration frequencies in a solid this discrepancy can be accounted using one dimensional model of monoatomic lattice Thermal Energy & Heat Capacity Debye Model Density of States According to Quantum Mechanics if a particle is constrained; • the energy of particle can only have special discrete energy values. • it cannot increase infinitely from one value to another. • it has to go up in steps. • These steps can be so small depending on the system that the energy can be considered as continuous. • This is the case of classical mechanics. • But on atomic scale the energy can only jump by a discrete amount from one value to another. Definite energy levels Steps get small Energy is continuous • In some cases, each particular energy level can be associated with more than one different state (or wavefunction ) • This energy level is said to be degenerate. ( ) • The density of states is the number of discrete states per unit energy interval, and so that the number of states between and d will be ( )d . There are two sets of waves for solution; • Running waves • Standing waves Running waves: 0 4 L 2 L 2 L 4 L 6 L k These allowed k wavenumbers corresponds to the running waves; all positive and negative values of k are allowed. By means of periodic boundary condition an integer Na 2 2 2 L Na p k pk p p k Na L Length of the 1D chain These allowed wavenumbers are uniformly distibuted in k at a density of R k between k and k+dk. running waves L R k dk dk 2 5 L Standing waves: 0 L 2 L k 3 L 6 L 7 L 4 L 0 3 L L 2 L In some cases it is more suitable to use standing waves,i.e. chain with fixed ends. Therefore we will have an integral number of half wavelengths in the chain; L n 2 2 n n ;k k k 2 2L L These are the allowed wavenumbers for standing waves; only positive values are allowed. 2 k p L for running waves k L p for standing waves These allowed k’s are uniformly distributed between k and k+dk at a density of S (k ) S (k )dk R k dk L dk L dk 2 DOS of standing wave DOS of running wave •The density of standing wave states is twice that of the running waves. •However in the case of standing waves only positive values are allowed •Then the total number of states for both running and standing waves will be the same in a range dk of the magnitude k •The standing waves have the same dispersion relation as running waves, and for a chain containing N atoms there are exactly N distinct states with k values in the range 0 to / a . The density of states per unit frequency range g(): • The number of modes with frequencies and +d will be g()d. • g() can be written in terms of S(k) and R(k). dR dn modes with frequency from to +d corresponds modes with wavenumber from k to k+dk dn S (k )dk g ( )d dn R (k )dk g ( )d; Choose standing waves to obtain g ( ) g ( ) S (k ) dk d Let’s remember dispertion relation for 1D monoatomic lattice 4K 2 ka sin m 2 2 d 2a dk 2 K ka cos m 2 K ka 2 sin m 2 g ( ) S (k ) 1 K ka a cos m 2 1 g ( ) S (k ) a m 1 K cos ka / 2 sin x cos x 1 cos x 1 sin x 2 2 2 1 g ( ) S (k ) a m K 1 ka 1 sin 2 2 4 4 ka 2 ka cos 1 sin 2 2 Multibly and divide Let’s remember: g ( ) S (k ) g ( ) 1 a 2 4 K 4 K 2 ka sin m m 2 1 L 2 2 a max 2 True density of states S (k )dk L dk L Na 4K ka 2 sin 2 m 2 4K 2 max m g ( ) g ( ) N m K 2N 2 max True density of states by means of above equation max K 2 m K m 2 1/ 2 constant density of states K True DOS(density of states) tends to infinity at max 2 , m since the group velocity d / dk goes to zero at this value of . Constant density of states can be obtained by ignoring the dispersion of sound at wavelengths comparable to atomic spacing. The energy of lattice vibrations will then be found by integrating the energy of single oscillator over the distribution of vibration frequencies. Thus 1 2 e 0 / kT g d 1 2N Mean energy of a harmonic oscillator 2 max for 1D 2 1/ 2 One can obtain same expression of g ( ) by means of using running waves. It should be better to find 3D DOS in order to compare the results with experiment. 3D DOS • Let’s do it first for 2D • Then for 3D. • Consider a crystal in the shape of 2D box with crystal lengths of L. ky y L 0 + - - + + L L L kx x Standing wave pattern for a 2D box Configuration in k-space •Let’s calculate the number of modes within a range of wavevector k. •Standing waves are choosen but running waves will lead same expressions. •Standing waves will be of the form U U 0 sin k x x sin k y y • Assuming the boundary conditions of •Vibration amplitude should vanish at edges of x 0; y 0; x L; y L Choosing p q kx ; ky L L positive integer ky y + L 0 - - + + - L L L kx x Standing wave pattern for a 2D box Configuration in k-space •The allowed k values lie on a square lattice of side positive quadrant of k-space. in /the L •These values will so be distributed uniformly with a density of 2 L / perunit area. • This result can be extended to 3D. L Octant of the crystal: kx,ky,kz(all have positive values) The number of standing waves; L 3 V 3 L 3 s k d k d k 3 d k 1 4 k 2 dk L / 8 V 1 3 s k d k 3 4 k 2 dk 8 2 Vk s k d 3k 2 dk 2 ky Vk 2 S k 2 2 3 L kz dk k kx 2 Vk • k is a new density of states defined as the number of 2 2 states per unit magnitude of in 3D.This eqn can be obtained by using running waves as well. • (frequency) space can be related to k-space: g d k dk g k dk d Let’s find C at low and high temperature by means of using the expression of g . High and Low Temperature Limits 3NkBT • Each of the 3N lattice modes of a crystal containing N atoms d C dT C 3NkB This result is true only ifT kB At low T’s only lattice modes having low frequencies can be excited from their ground states; long Low frequency sound waves vs k 0 k a vs k k 1 dk 1 vs k vs d vs 2 V 2 vs 1 g 2 2 vs and Vk 2 dk g 2 2 d at low T depends on the direction and there are two transverse, one vs longitudinal acoustic branch: V2 1 V2 1 2 g g 3 2 3 2 3 2 vs 2 vL vT Velocities of sound in longitudinal and transverse direction 1 2 e 0 / kT 1 2 e 0 g d 1 / kT Zero point energy = z 2 V 1 2 2 3 3 d 1 2 vL vT x V 1 2 3 z 2 3 3 / kT d 2 vL vT 0 e 1 3 V 1 2 k BT 4 z 2 3 3 3 2 vL vT 15 4 e 0 e 3 / kT 0 / kT 3 1 1 d d 0 k BT 3 k BT 3 x kT B dx x e 1 k BT k BT x d k BT dx 4 x3 0 e x 1dx 4 15 1 2 kBT d 2 2 Cv V kB 3 3 dT 15 v v L T 3 at low temperatures How good is the Debye approximation at low T? 1 d 2 2 k BT 2 Cv V kB 3 3 dT 15 v v L T The lattice heat capacity of solids thus varies as T 3at low temperatures; this is referred to as the Debye T 3 law. Figure illustrates the excellent agreement of this prediction with experiment for a non-magnetic insulator. The heat capacity vanishes more slowly than the exponential behaviour of a single harmonic oscillator because the vibration spectrum extends down to zero frequency. 3 The Debye interpolation scheme The calculation of g ( ) is a very heavy calculation for 3D, so it must be calculated numerically. Debye obtained a good approximation to the resulting heat capacity by neglecting the dispersion of the acoustic waves, i.e. assuming s k for arbitrary wavenumber. In a one dimensional crystal this is equivalent to taking g ( ) as given by the broken line of density of states figure rather than full curve. Debye’s approximation gives the correct answer in either the high and low temperature limits, and the language associated with it is still widely used today. The Debye approximation has two main steps: 1. Approximate the dispersion relation of any branch by a linear extrapolation of the small k behaviour: Einstein approximation to the dispersion Debye approximation to the dispersion vk Debye cut-off frequency D 2. Ensure the correct number of modes by imposing a cutoff frequency D, above which there are no modes. The cut-off freqency is chosen to make the total number of lattice modes correct. Since there are 3N lattice vibration modes in a crystal having N atoms, we choose D so that D V2 1 2 g ( ) ( 3 3) 2 g ( )d 3N 2 vL vT 0 V 1 2 ( )D3 3 N 2 3 3 6 vL vT g ( ) 9N D3 V 1 2 D 2 ( ) d 3N 2 2 vL3 vT3 0 V 1 2 3N 9N ( ) 3 2 2 vL3 vT3 D3 D3 2 g ( ) / 2 The lattice vibration energy of E ( 0 becomes and, 9N E 3 D 1 / kBT )g ( )d 2 e 1 D D 3 1 9 N 3 2 0 ( 2 e / kBT 1) d D3 0 2 d 0 e / kBT 1d D 9 9N E N D 3 8 D D 3d e 0 / k BT 1 First term is the estimate of the zero point energy, and all T dependence is in the second term. The heat capacity is obtained by differentiating above eqn wrt temperature. C The heat capacity is 9 9N E N D 3 8 D D 0 d e / k BT 1 3 dE dT dE 9 N CD 3 dT D D 0 4 e / k BT d 2 2 kBT e / kBT 1 2 Let’s convert this complicated integral into an expression for the specific heat changing variables to x k BT d kT dx and define the Debye temperature D D kB kT x The Debye prediction for lattice specific heat dE 9 N kBT kBT CD 3 2 dT D kBT 4 T CD 9 Nk B D where D 2 3 /T D D kB 0 D / T 0 x 4e x e 1 x x 4e x e x 1 2 2 dx dx How does C D limit at high and low temperatures? High temperature x is always small T D x2 x3 e 1 x 2! 3! x x 4 (1 x) 2 x 2 2 2 x x 1 x 1 e 1 x 4e x T x 4 (1 x) T D CD 9 Nk B D 3 /T D 0 x 2 dx 3Nk B How does C D limit at high and low temperatures? Low temperature T D For low temperature the upper limit of the integral is infinite; the integral is then a known integral of . 4 4 /15 T T D CD 9 Nk B D 3 /T D 0 x 4e x e x 1 2 dx We obtain the Debye T 3 law in the form CD 12 Nk B T 5 D 4 3 Lattice heat capacity due to Debye interpolation scheme T CD 9 Nk B Figure shows the heat capacity D 3 D / T 0 x 4e x e x between the two limits of high and low T as predicted by the Debye interpolation formula. 1 2 dx C 3 Nk B T 1 Because it is exact in both high and low T limits the Debye formula gives quite a good representation of the heat capacity of most solids, even though the actual phonon-density of states curve may differ appreciably from the Debye assumption. Lattice heat capacity of a solid as predicted by the Debye interpolation scheme 1 T / D Debye frequency and Debye temperature scale with the velocity of sound in the solid. So solids with low densities and large elastic moduli have high D . Values of Dfor various solids is given in table. Debye energy D can be used to estimate the maximum phonon energy in a solid. Solid D (K ) Ar Na Cs Fe Cu Pb C KCl 93 158 38 457 343 105 2230 235