14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
10.1146/annurev.physiol.65.092101.142213
Annu. Rev. Physiol. 2003. 65:383–400
doi: 10.1146/annurev.physiol.65.092101.142213
c 2003 by Annual Reviews. All rights reserved
Copyright °
First published online as a Review in Advance on October 4, 2002
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
INSIGHTS INTO THE REGULATION OF GASTRIC
ACID SECRETION THROUGH ANALYSIS OF
GENETICALLY ENGINEERED MICE
Linda C. Samuelson and Karen L. Hinkle
Department of Physiology, The University of Michigan, Ann Arbor, Michigan,
48109-0622; e-mail: lcsam@umich.edu; khinkle@umich.edu
Key Words transgenic and knockout mice, acid secretagogues, gastrin, histamine,
parietal cells, enterochromaffin-like cells, gastric epithelium
■ Abstract The regulation of acid secretion in the stomach involves a complex
network of factors that stimulate secretion in response to the ingestion of a meal
and maintain homeostasis of gastric pH. Genetically engineered mouse models have
provided a new opportunity to investigate the importance and function of specific
molecules and pathways involved in the regulation of acid secretion. Mouse mutants
with disruptions in the three major stimulatory pathways for acid secretion in parietal
cells, gastrin, histamine, and acetylcholine, have been generated. Disruption of the
gastrin pathway results in a major impairment in both basal and induced acid secretion. Histamine and acetylcholine pathway mutants also have significant alterations in
acid secretion, although the impairment does not appear to be as severe as in gastrin
pathway mutants, perhaps due in part to the hypergastrinemia that occurs. Mice with
a disruption in the somatostatin pathway have increased gastric acid secretion, which
confirms an important negative regulatory role for this factor. This review discusses
these genetically engineered mouse models, as well as others, that provide insight into
the complex regulation of in vivo gastric acid secretion. The regulation of growth and
cellular morphology of the stomach in these mouse models is also presented. In addition, transgene promoters that are expressed in the gastric epithelium are discussed
because these promoters will be important tools to alter cellular physiology in new
mouse models in the future.
INTRODUCTION
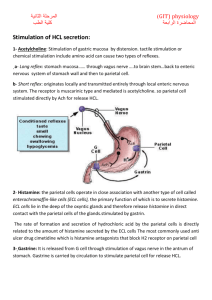
A complex network involving endocrine, neural, and paracrine stimulation of epithelial cells in the gastric mucosa exists to regulate gastric acid secretion (Figure 1).
Acid is secreted from the parietal cell, which is one of the most abundant cell
types in the corpus of the stomach. The three major factors that activate the
parietal cell to secrete acid are gastrin, histamine, and acetylcholine (ACh) (1).
Gastrin primarily activates the parietal cell through an indirect pathway involving
0066-4278/03/0315-0383$14.00
383
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
384
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
Figure 1 Model for the regulation of gastric acid secretion. Shown are a parietal cell,
an ECL cell, a D cell, and many of the important regulators of acid secretion. Parietal
cells are stimulated by three main agonists: gastrin, histamine, and ACh. Gastrin binds
to CCKB receptors on the parietal cell to evoke an increase in intracellular calcium;
histamine binds to H2 receptors, which primarily signal through increased cAMP;
ACh binds to M3 receptors to stimulate an increase in intracellular calcium. Parietal
cell stimulation results in movement of H+/K+-ATPase pumps to the apical membrane
to secrete acid. Histamine is released from ECL cells in response to gastrin stimulation
as well as neuronal stimulation via PACAP. Somatostatin (Sst) released from D cells
inhibits acid secretion by reducing ECL cell histamine release, blocking gastrin release,
and directly inhibiting parietal cell acid secretion.
the release of histamine from enterochromaffin-like (ECL) cells. Histamine acts
in a paracrine manner to stimulate parietal cells to secrete acid by binding to histamine 2 (H2) receptors. Gastrin can also stimulate parietal cells directly by binding
to gastrin/cholecystokinin-B (CCKB) receptors, although the importance of direct
versus indirect stimulation is not fully understood. The third major stimulatory
pathway involves neural activation with release of ACh, which binds to muscarinic
3 (M3) receptors on the parietal cell. Neural stimulation can also activate ECL
cell histamine release, most likely through pituitary adenylate cyclase-activating
polypeptide (PACAP) binding to PACAP-1 receptors on the ECL cell (2, 3). The
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
ACID SECRETION IN MOUSE MUTANTS
385
stimulation of histamine release from ECL cells by gastrin and ACh means that
these pathways are often activated in concert to stimulate secretion from the parietal
cell.
In parietal cells, CCKB and M3 receptors couple to Gq, which upon stimulation
activates phospholipase C to induce an increase in inositol trisphosphate and the
release of intracellular calcium (4). The H2 receptor in these cells couples primarily
to Gs, which activates adenylate cyclase and evokes an increase in cAMP (5),
although it has been shown in some species also to be coupled to an intracellular
calcium pathway (6). There is evidence in vitro that increased levels of both cAMP
and intracellular calcium are required for stimulated acid secretion (7, 8).
Somatostatin is a paracrine regulator of acid secretion that likely acts as a
physiological inhibitor, operating at several different levels. It is synthesized in
and secreted from D cells in the acid-secreting corpus of the stomach and in the
antrum, where gastrin-producing G cells are located. Somatostatin release has
been associated with the inhibition of gastrin synthesis and secretion (9, 10), the
inhibition of histamine release by ECL cells (11, 12), and direct inhibition of parietal cell acid secretion (13, 14; Figure 1). Molecular and pharmacological studies
suggest that the somatostatin subtype 2 (sst2) receptor is the predominant receptor
regulating ECL cell histamine release (12, 15) and acid secretion (14, 16–20).
The epithelial cells of the gastric mucosa are organized into gastric glands,
which are the functional units of the gastric acid secretory system. In the corpus,
each gland is made up of four characteristic regions: (a) the pit region, which
lies at the top of the gland near the lumen and contains primarily mucous cells;
(b) the isthmus, which contains immature progenitor cells; (c) the neck, which
also contains mucous cells; and (d ) the base, which lies closest to the basement
membrane and contains ECL cells and chief cells. Parietal cells are located in all
regions of the gland, although they are most dense in the central portion, including
the isthmus and neck. The progenitor stem cells that reside in the isthmus give rise
to all the epithelial cell types in the gastric gland (21).
Parietal cells undergo dynamic and distinct morphological changes when stimulated by an acid secretagogue. In the unstimulated state, parietal cells contain
abundant intracellular membrane compartments, known as tubulovesicles, that sequester H+/K+-ATPase pumps beneath the cell surface. Upon stimulation, these
tubulovesicles fuse with the apical (canalicular) membrane of the parietal cell,
exposing H+/K+-ATPase pumps to the lumen and thus enabling acid secretion to
take place (22, 23). In the gastric mucosa, the H+/K+-ATPase α- and β-subunits are
expressed specifically in the parietal cell and are therefore used as markers for this
cell type. Cessation of acid secretion occurs upon internalization of H+/K+-ATPase
and the re-establishment of the intracellular tubulovesicles (22, 23).
The mouse is an excellent animal model to merge molecular, cellular, and integrative biology for the study of acid secretion. One strength of the mouse as an
experimental model is the potential for genome engineering by gene targeting in
embryonic stem (ES) cells or by conventional transgenesis. Gene targeting allows
the replacement of the normal gene with a mutant gene construct through homologous recombination, thus allowing the analysis of loss-of-function mutations. In
14 Jan 2003
14:2
386
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
contrast, conventional mouse transgenics have a gene construct that is randomly
integrated in the mouse genome. Because the normal mouse genome is retained,
conventional transgenic approaches are suitable only for dominant phenotypes.
This review focuses on the physiology of gastric acid secretion in genetically
engineered mouse models. Several transgenic and knockout mouse models with
alterations in gastric acid secretion have been generated over the past several years
(Table 1). Because acid secretion is not a vital function, these mutants are viable
and fertile, allowing for the careful examination of gastric morphology and acid
TABLE 1 Genetically engineered mouse models with alterations in gastric acid secretion
Mouse model
Acid
Gastrina
Gastrin pathway
Gastrin KO
Reduced
Absent
Reduced
High
Increased
High
Alteredc
High
Reduced
High
Acetylcholine pathway
M3 receptor KO
Reduced
Somatostatin pathway
Receptor 2 KO
Increased
CCK-B
receptor KO
Gastrin
overexpression
Histamine pathway
H2 receptor KO
HDC KO
Method
References
Targeting
(24, 25)
Targeting
(26, 27)
Transgenic
(32–34)
Hyperplasia: increased
parietal and ECL cells
Hyperplasia: increased
parietal and ECL cells
Targeting
(38)
Targeting
(39)
High
NDd
Targeting
(43)
Normal
NDd
Targeting
(45)
Abnormal parietal cells;
hyperplasia
Abnormal parietal cells;
hyperplasia
Mucous cell hyperplasia
Targeting
(50)
Targeting
(35)
Transgenic
(56)
Targeting
(53, 54)
Targeting
(55)
Parietal cells-acid secretory machinery
H/K-ATPase α KO
Absent
High
H/K-ATPase β KO
Absent
High
H/K-ATPase
β mutant
NHE2 KO
Increased
ND
Absent
ND
Absent
or low
High
KvLQT1 KO
a
Thinner mucosa: altered
parietal and ECL cells
Thinner mucosa: fewer
parietal and ECL cells
Hyperplasia: increased
parietal and ECL cells;
Older mice: atrophy with
loss of parietal cells
Reduced parietal and chief
cells; surface mucous cell
hyperplasia
Surface mucous cell
hyperplasia
Circulating plasma gastrin levels in the various mutants relative to wild type mice are indicated.
b
c
Gastric mucosal
cell changesb
See text for a description of acid secretory and mucosal cell changes in the various mutants.
H2 receptor-deficient mice are characterized by normal basal acid, and lack of responsiveness to histamine and gastrin.
d
ND = not determined.
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
ACID SECRETION IN MOUSE MUTANTS
387
secretory function. These genetically engineered mouse models have provided an
opportunity to re-evaluate the importance and function of specific molecules and
pathways for the in vivo regulation of acid secretion and the growth and development of the acid secretory system.
GASTRIN PATHWAY
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
Gastrin Loss-of-Function
Gastrin is the principal hormonal inducer of gastric acid secretion. Mice with
mutations in the genes encoding gastrin (24, 25) and the gastrin (CCKB) receptor
(26, 27) have been generated by gene targeting in ES cells. These null mutations
have allowed the further investigation of the importance of gastrin for the overall
function of the gastric acid secretory system. Because an extensive body of research
indicated that gastrin is a physiological regulator of acid secretion (28), it was expected that the loss of gastrin signaling would result in decreased acid secretion.
However, the extent of the impairment in acid secretion observed in these mutants
was greater than anticipated. Gastrin mutants had marked reductions in both basal
and induced acid secretion. Acid secretory function was measured in a number of
different ways in these mutants with similar results. Analysis of acid secretion in
gastrin-deficient mice was performed by perfusion in anesthetized mice with and
without stimulation with acid secretogogues (25). In the CCKB receptor-deficient
mutants, acid secretion was measured by using the pyloric ligation method (26).
Measurement of resting gastric pH was performed in both ligand- and receptordeficient mice (24, 27). In each case, basal gastric acid levels were significantly
reduced in the gastrin pathway mutants. Stimulated acid secretion was also severely
impaired in gastrin-deficient mice, as acute induction with histamine, carbachol,
or gastrin did not increase acid (25). This finding was surprising because previous
experimental models in which gastrin-induced acid secretion was acutely blocked
with a gastrin immunoneutralizing antibody or CCKB receptor antagonists were
still responsive to other agonists (29, 30). The lack of agonist-stimulated acid secretion in gastrin-deficient mice is unique and suggests a fundamental requirement of
gastrin for acid secretion and/or for the development of the acid secretory system.
Gastrin replacement for 6 days by continuous perfusion using osmotic minipumps
was able to partially restore acid secretion in gastrin-deficient mice (25). Thus
the cellular components of the acid secretory system are capable of secreting acid
once gastrin is provided. The lack of an acid secretory response to acute gastrin
treatment suggests that the gastrin-induced repair requires a longer period of time.
There were significant changes to both parietal and ECL cells in gastrin pathway mutants. Thus the defect in basal and induced acid secretion was likely due
to the loss of gastrin stimulation of both of these cell types. There was a thinning
of the gastric mucosa in CCKB receptor-deficient mice (26), which is consistent
with the known trophic effect of gastrin on the gastric mucosa (28, 31). In addition,
the number of parietal and ECL cells was reduced in both gastrin-deficient and
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
388
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
CCKB receptor-deficient mutants (24–27). Furthermore, differentiated markers of
ECL function were reduced, including gastric histamine content, as well as the
expression of chromogranin A (CgA) and the histamine biosynthetic enzyme histidine decarboxylase (HDC) (24, 25). Therefore, reductions in both histamine and
gastrin stimulation of the parietal cell are characteristic of gastrin pathway mutants. It is not clear what aspects of the phenotype result from loss of direct gastrin
stimulation of the parietal cell versus loss of histamine stimulation of the parietal
cell. The lack of response to histamine stimulation in gastrin-deficient mice (25)
suggests either that the acid secretory machinery is not fully functional in these
mice or that histamine signaling alone is not sufficient to induce acid secretion
in vivo.
Gastrin Overexpression
It is useful to compare the phenotypes of gastrin overexpression mouse models to
gastrin loss-of-function models to further investigate in vivo gastrin function. The
INS-GAS transgenic mouse, in which gastrin expression is driven by the insulin
promoter, exhibits a twofold increase in circulating amidated gastrin, which results
from the expression of a human gastrin transgene in pancreatic β-cells (32). At
4 months of age, basal acid levels are increased approximately threefold in this
mutant, consistent with the idea that gastrin is a key inducer of the acid secretory
system (33). There is also an increase in the number of parietal cells observed at
this age. In general, mouse models with increased circulating gastrin exhibit gastric
mucosal cell hyperplasia with increased parietal and ECL cells when examined
at younger ages (Table 1). Interestingly, the increased acid secretion seen in the
INS-GAS transgenics was lost as the mice aged, because of a developing gastric
atrophy characterized by the loss of parietal cells and mucous cell hyperplasia (33).
Similar gastric histopathology was observed in a second gastrin transgenic mouse
model that exhibited a sixfold elevation of amidated gastrin (34). The changes that
occurred with aging in these transgenics underscore the importance of examining
mice at various ages for analysis of gastric physiology. Indeed, age-related changes
in acid secretion and mucosal cell histology have been detected in other studies
for both wild-type and mutant mice (33–35).
HISTAMINE PATHWAY
Histamine and Acid Secretion
The important contribution of histamine to the regulation of gastric acid secretion
has been demonstrated by the effectiveness of H2 receptor antagonists to block acid
(36, 37). Moreover, histamine receptor antagonists have also been shown to block
responses to gastrin and carbachol, suggesting that histamine is the most significant
inducer of acid secretion (37). Because histamine is such a potent inducer of acid
and also may provide a necessary parietal cell cAMP signal, it seems likely that
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
ACID SECRETION IN MOUSE MUTANTS
389
both basal and induced acid secretion would be impaired in histamine pathway
mouse mutants. However, it was surprising that histamine pathway disruption
by targeted mutagenesis showed a less severe acid secretory impairment than in
gastrin pathway mutants.
Two different histamine pathway mouse mutants have been generated: an H2
receptor-deficient (38) and an HDC-deficient strain (39). These two mutants share
many features, including measurable basal acid secretion, normal responses to
carbachol, and loss of responsiveness to gastrin. Hypergastrinemia was also observed in both mutants, which is a characteristic response to low acid secretion.
Circulating gastrin levels are regulated by acid content in the stomach by a classic
feedback mechanism in which gastrin synthesis and secretion are increased when
gastric pH is high. It has not yet been determined if the hypergastrinemia in the
histamine pathway mutants contributes to the normalization of acid levels, but this
could be tested by treatment with a gastrin receptor antagonist. The lack of responsiveness to gastrin stimulation suggests that histamine signaling may be required
in vivo for gastrin responses to be observed. Because increased acid secretion in
response to gastrin stimulation has been observed in overexpression transgenics
with similar levels of hypergastrinemia (34), it is unlikely that the lack of gastrin
responsiveness in the histamine pathway mutants is due to maximal stimulation
or desensitization of gastrin stimulation of the parietal cells. The observation that
histamine is not required for basal or carbachol-induced acid secretion suggests
that other pathways can also increase parietal cell cAMP or that a cAMP signal is
not required in vivo.
The H2 receptor and HDC mutants would be expected to similarly disrupt parietal cell histamine signaling to the parietal cell, which is thought to be H2 receptor
mediated. Indeed, these two mutants had many similarities, as indicated above.
However, it is noteworthy that these two mutants also showed important differences. Measurement of basal acid secretion demonstrated a modest but significant
reduction in the HDC-deficient mouse compared with wild-type controls (39). In
contrast, H2 receptor-deficient mice had normal basal acid secretion (38). These results were confirmed in a preliminary side-by-side comparison of H2 receptor- and
HDC-deficient mutants showing lower acid in HDC-deficient mutants but normal
acid in H2 receptor-deficient mice (40). Non-H2 receptor-mediated effects could
contribute to the difference in basal acid. Loss of HDC would affect histamine
production throughout the body, and all histamine signaling would be disrupted,
including H1, H2, and H3 receptor-mediated effects; in contrast, only H2 receptormediated processes would be blocked in the H2 receptor mutant mouse. There is
some evidence that regulation of acid secretion may include H3 receptor-mediated
processes. For example, in a study using isolated mouse stomachs, the H3 receptor
antagonist thioperamide was shown to increase somatostatin, decrease histamine,
and decrease acid in a dose-dependent manner (41). Thus it is possible that somatostatin may be upregulated in the HDC mutant through this pathway, which
would result in the reduced acid secretion. This will be an interesting hypothesis
to test in these mice.
14 Jan 2003
14:2
390
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
Comparison of Histamine Pathway and Gastrin
Pathway Mutants
Comparison of the phenotype of gastrin pathway mouse mutants with the phenotype of histamine pathway mutants provides insight into the importance of direct
gastrin stimulation versus indirect gastrin stimulation of the parietal cell. Mice
with gastrin pathway mutations would be expected to lose both direct and indirect
stimulation, whereas histamine pathway mutants would maintain direct gastrin
effects on the parietal cell (Figure 1). Indeed, the data show that the gastrin pathway mutants have a more severe phenotype. Both basal and induced acid secretion
were severely impaired in gastrin-deficient mice (25), whereas histamine pathway mutants had near normal basal secretion (38, 39). Thus gastrin must at least
partially stimulate the gastric acid secretory system via a histamine-independent
pathway, suggesting some component of direct stimulation of the parietal cell. Indeed, parietal cells are known to have CCKB receptors, and isolated parietal cells
can respond to gastrin in vitro (42). On the other hand, it has been determined that
H2 receptor antagonists are effective in vivo acid blockers, even upon stimulation
with gastrin (37). Thus it is evident that in vivo acid secretion involves both gastrin
and histamine stimulation of the parietal cell.
Gastric Mucosal Hypertrophy
Another common phenotype in the histamine pathway mutants is mucosal hypertrophy. In the H2 receptor-deficient mouse, stomach wet weight was significantly
increased, with increased numbers of parietal and ECL cells (38). In addition, there
was an increase in cellular proliferation, as measured by 5-bromo-20 -deoxyuridine
incorporation (38). The hypertrophy is apparent in the H2 receptor mutant even at
16 weeks of age and leads to grossly enlarged mucosal folds. Hypertrophy was
not observed in the original HDC-deficient mouse study (39). However, a preliminary study by Okabe et al. (40) reported hypertrophy, which was associated
with increased parietal and ECL cell numbers, in both HDC- and H2 receptordeficient mutant mice. The basis for the discrepancy between the two studies is
not clear, but it may have to do with the age of the animals that were studied.
Hypergastrinemia in both histamine pathway mutants is the likely explanation for
the hypertrophy, as treatment with the gastrin receptor antagonist YM-022 resulted
in reduced hyperplasia (40).
ACETYLCHOLINE PATHWAY
To investigate the role of ACh in the regulation of acid secretion, an M3 receptordeficient mouse was created (43). In this preliminary report, an elevated resting
intragastric pH indicated defects in basal acid secretion. However, stimulated acid
secretion was observed in M3 receptor-deficient mice after treatment with ACh, histamine, and gastrin, demonstrating that the acid secretory system was responsive.
The response to ACh appeared to operate through a histamine pathway because
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
ACID SECRETION IN MOUSE MUTANTS
391
the H2 receptor antagonist famotidine blocked the response. Hypergastrinemia was
observed in the mutant, suggesting that feedback mechanisms were operating in
this mutant to compensate for lower parietal cell stimulation by increasing gastrin
levels. Gastric mucosal morphology has not yet been reported for this mutant, but
it is expected that the hypergastrinemia would lead to hypertrophy similar to that
described for the histamine pathway mutants.
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
SOMATOSTATIN PATHWAY
Somatostatin is thought to be a paracrine inhibitor of acid secretion that operates
at many levels (Figure 1). Somatostatin regulation is complicated; there are at least
five receptor subtypes. Molecular and pharmacologic studies suggest that the sst2
receptor is the predominant receptor regulating acid secretion (12, 14–20). The
production of a sst2 receptor-deficient mouse model by Zheng et al. (44) enabled
Martinez et al. (45) to investigate the consequences of the loss of all of the isoforms
of this receptor for gastric acid secretion. Analysis of acid secretion in the mutant
supports the conclusion that somatostatin, acting through the sst2 receptor, is an
inhibitor of gastric acid secretion.
Measurement of resting intragastric pH showed no differences between sst2
receptor-deficient mice and wild-type controls, which suggests that this receptor
does not mediate tonic inhibition of acid secretion by somatostatin. However, a very
significant phenotype was uncovered when studies were performed with urethane
anesthesia, which has the interesting property of stimulating gastric somatostatin
release (46, 47). Acid secretion in anesthetized wild-type mice was approximately
10-fold lower than in sst2 receptor-deficient mice (45). This result suggests that
the sst2 receptor normally mediates inhibition of acid secretion when somatostatin
levels are increased. Interestingly, there were no changes in circulating gastrin
levels, suggesting that somatostatin inhibition of gastrin release does not occur
through a sst2 receptor-mediated pathway. There was no examination of gastric
morphology in this mutant, so it is not known if loss of the sst2 receptor affects
the cellular composition of the mucosa. It will be important to complete the morphological analysis of the sst2 receptor-deficient strain, as well as to examine the
recently generated somatostatin-deficient mice (48, 49) to more fully understand
the consequences of the loss of somatostatin for gastric acid physiology.
PARIETAL CELL ACID SECRETORY MACHINERY
Genetic engineering in the mouse has also allowed the investigation of the roles
of several ion transporters and channels in gastric acid secretion. For example,
it has been shown by gene targeting experiments in the mouse that both the αand β-subunits of the H+/K+-ATPase are essential for normal acid secretion from
the parietal cell (35, 50, 51). The acid secretory phenotypes of the two mutants
are similar. The α- and β-subunit-deficient mutants were achlorhydric, indicating that each of these subunits is required for acid secretion from the parietal
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
392
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
cell. In addition, plasma gastrin levels were markedly elevated as a result of the
low acid levels in these mice. Major alterations in parietal cell morphology were
also noted in both the α- and β-subunit-deficient mouse models, including dilated canaliculi and changes in canalicular microvilli. However, differences were
noted between the two H+/K+-ATPase subunit mutants in the cellular makeup of
the gastric glands. In particular, whereas the H+/K+-ATPase β-subunit-deficient
mouse showed significant decreases in chief and parietal cells and increases in pit
and neck mucous cells, there were no changes detected in the numbers of these
cell types in the α-subunit-deficient mouse. This suggests that the β-subunit of
H+/K+-ATPase is required for the normal maintenance and distribution of several
cell types within the gastric glands, whereas the α-subunit seems to be important
for the normal morphology of only parietal cells.
In the H+/K+-ATPase α- and β-subunit mutants, it was not known whether
abnormal parietal cell morphology and hyperplasia were the result of increased
gastrin levels, loss of acid, or some other factor. To address which aspects of
the phenotype of the H+/K+-ATPase β-subunit-deficient strain might be due to
high gastrin levels, these mice were crossed with gastrin-deficient mice (52). The
H+/K+-ATPase β-subunit/gastrin-deficient double mutant mice showed abnormal
parietal cell morphology, suggesting that this aspect of the phenotype of H+/K+ATPase β-subunit mutant mice was not due to high gastrin levels. However, the
hyperplasia and increased numbers of pit and neck cells observed in H+/K+ATPase β-subunit mice were not apparent in the double mutant, suggesting that
these aspects of the original mutant resulted from elevated levels of gastrin. In any
case, analysis of H+/K+-ATPase α- and β-subunit-deficient mice suggests that the
proton pump is not only essential for acid secretion but is also required for normal
parietal cell morphology.
Other mouse models have been engineered with disruptions in ion exchange,
including Na+/H+ exchanger isoform 2–deficient (NHE2) (53, 54) and KvLQT1
voltage-gated potassium channel–deficient (55) strains. There are many similarities between these strains and the H+/K+-ATPase α- and β-subunit-deficient mice.
Both NHE2, which normally is present on the basolateral membrane of parietal
cells, and KvLQT1, which previously was unknown to play a role in gastric function, were shown to be essential for normal acid secretion. As previously seen in
other hypochlorhydric models, gastrin levels were elevated in both the KvLQT1
and NHE2 mutant mice, resulting in hyperplasia of mucous and undifferentiated epithelial cells. Similar to the H+/K+-ATPase β-subunit-deficient mice, parietal and chief cells were reduced in both NHE2- and KvLQT1-deficient mice.
In addition, parietal cells in these strains were abnormal and were characterized
by an increase in vacuolization, as was also seen in H+/K+-ATPase α- and βsubunit-deficient mice. The striking similarities between the NHE2, KvLQT1,
and H+/K+-ATPase-deficient mouse models suggest that the structural aspects of
the ion exchanger proteins may not be essential for normal acid secretion and mucosal morphology, but rather the disruption of proper ion exchange or membrane
gradients is responsible for the alterations in gastric mucosal morphology seen in
these mutants.
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
ACID SECRETION IN MOUSE MUTANTS
393
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
TRANSGENE EXPRESSION IN THE GASTRIC MUCOSA
Several transgenic mouse experiments have been performed to alter the development or physiology of the gastric mucosa. Transgene promoters have been used to
drive cell-specific expression in the gastric mucosa, including parietal cells, neuroendocine cells, G cells, and the squamous epithelium of the forestomach (Table 2;
described below). In the future, it will be advantageous to define promoters to direct transgene expression to additional cell types in the gastric mucosa, including
mucous pit and neck cells, chief cells, D cells, and ECL cells. The ability to manipulate the physiology of these cells through transgene expression is a useful tool
to understand the complex interactions that occur in the gastric mucosa to regulate
acid secretion.
Parietal Cell Transgenics
Recently, an elegant transgenic mouse model was described by Courtois-Coutry
et al. (56) in which parietal cells were shown to constitutively secrete acid. In this
study, a transgene was constructed that contained the H+/K+-ATPase β-subunit
with a mutation in the internalization signal; this mutation resulted in the inability
of H+/K+-ATPase to be resequestered from the apical membrane and led to constitutive acid secretion. The cytomegalovirus (CMV) promoter was used to drive
expression of the H+/K+-ATPase β mutant transgene. Although this is a strong
constitutive promoter with expression in a number of tissues, functional changes
are limited to cells that express H+/K+-ATPase, including parietal cells in the stomach (56) and renal tubule epithelial cells (57). The H+/K+-ATPase β transgenic
mice were shown to hypersecrete acid, and over the course of several months they
TABLE 2 Cell-specific expression of transgene promoters in the gastric mucosa
Promoter
Cell-specific
expression
DNA fragmenta
Species
References
H/K-ATPase
β subunit
Parietal cell
−1035 bp to +24 bp
−13.5 kb to −29 bp
Mouse
Mouse
(58–60, 62)
(61)
Chromogranin A
ECL and other
neuroendocrine
cells
−4.8 kb to +42 bp
Mouse
(68)
Gastrin
G cell
−450 bp to +550
(rat exon 1) and 4 kb
human exons 2 and 3
Rat/human
(32, 69)
ADAb
Forestomach
squamous
epithelium
−6.4 kb to +90
−4.4 kb to −3.3 kb and
−750 bp to +90
Mouse
Mouse
(70)
(71)
a
Genomic fragments used to construct the transgenes are indicated. Numbers refer to the start of transcription (+1).
b
ADA, adenosine deaminase.
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
394
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
developed gastric ulcers and a hypertrophic gastropathy resembling Menetrier’s
disease (56). Gastrin levels were not measured in this study, but the increased acid
would be expected to result in reduced circulating gastrin.
The best-characterized transgene promoter for expression in the gastric mucosa
is the mouse H+/K+-ATPase β-subunit promoter. This promoter has been used in
a number of different experiments with transgene expression directed to parietal
cells of transgenic mice (58–62). Most studies have used an ∼1-kb mouse H+/
K+-ATPase β-subunit promoter fragment containing sequences extending from
−1035 bp to +24 bp. Transcription from this promoter was specific for the parietal cell lineage, as transgene expression was shown to be absent in both precursors
and differentiated members of the mucous pit and chief cell lineages. Moreover,
transgene expression was observed in the majority of parietal cells, as detected
by expression of the H+/K+-ATPase marker. In one study, the H+/K+-ATPase
β transgene was expressed in >95% of the parietal cells in three of four transgenic lines that were generated (58), which demonstrates the effectiveness of this
promoter for parietal cell expression. Although the transgenic mouse experiments
that used this promoter in the past focused on developmental questions, it will be a
powerful promoter for future experiments in which parietal cell physiology can be
manipulated.
Chromogranin A Promoter and ECL Cells
Chromogranin A (CgA) is involved in processing and/or sorting of proteins in secretory granules in neuroendocrine cells. A recent report described neuroendocrine
expression of a luciferase reporter gene in transgenic mice using the mouse CgA
promoter (63). In the stomach, the transgene was predominantly expressed in ECL
cells, which are the most abundant neuroendocrine cell types in the mouse corpus. Expression in some D cells and G cells was also observed. In addition to
this pattern of expression in the stomach, the transgene was expressed in other
tissues known to express endogenous CgA, including intestine, adrenal, pancreas,
and brain. In general, transgene expression was selective for the neuroendocrine
system and was similar to endogenous CgA expression. In gastric ECL cells, CgA
secretion and gene expression are regulated by gastrin (64–68), and accordingly
the CgA transgene promoter was also shown to be regulated by gastrin (63). With
omeprazole treatment, which blocks H+/K+-ATPase and normally induces hypergastrinemia, there was a fourfold increase in transgene expression. Although
the CgA promoter fragment defined in this study will be a powerful tool for manipulating neuroendocrine cells in future studies of gastric physiology, it would
also be desirable to define ECL cell– and D cell–specific transgene promoters to
independently manipulate these cell types.
Gastrin Promoter and G Cells
Transgene expression in G cells was achieved by creating a rat/human gastrin gene
chimera (rGAS-hGAS; 32). A 1-kb fragment containing the rat gastrin promoter
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
ACID SECRETION IN MOUSE MUTANTS
395
and first exon was fused to a 4-kb fragment containing human gastrin exons 2
and 3. In the stomach, this transgene was expressed specifically in the G cells in
the antrum. The pattern of transgene expression was similar to endogenous gastrin
gene expression, with the exception of higher transgene expression observed in the
duodenum. Two other gastrin transgenes were examined at the same time: rGAShGH, which contains the 1-kb rat promoter fused to a human growth hormone
reporter, and hGAS-hGAS, which contains a 2-kb human gastrin promoter linked
to the 4-kb human gastrin segment (32). Neither of these transgenes was expressed
in the stomach, demonstrating that both the rat and human gastrin segments are
required for G cell–specific expression in these transgenics. A derivative of the
chimeric transgene was also used in a recent study by Zhukova et al. (69) in which
a human insulin gene was inserted into the rat/human clone (Gas-Ins). Similar
to the results of Wang et al. (32), gastric antral–specific transgene expression in
G cells was detected, although in contrast to the previous study, no expression was
detected in duodenum. This chimeric sequence will be useful for directing further
transgenes into gastric G cells. However, better definition of the specific rat and
human gastrin sequences required for proper expression would make this a more
useful system.
ADA Promoter and Forestomach
An adenosine deaminase (ADA) promoter element has been described to direct high-level transgene expression to the stratified squamous epithelium in the
forestomach of the mouse (70, 71). ADA, a purine catabolic enzyme, is ubiquitously expressed, but the level of expression varies markedly among different
tissues. In mice, the highest levels of ADA occur in the gastrointestinal tract, including the squamous epithelium that lines the tongue, esophagus, and forestomach,
where the enzyme can account for as much as 20% of soluble protein (72). Initially,
transgenic mouse studies showed high expression with a 6.4-kb promoter fragment
(70). This was followed up with smaller constructs, leading to the identification
of a 1.1-kb upstream flanking sequence that is required for high-level expression
when paired with the natural promoter. Although this promoter was used for directing expression of a reporter gene, it would be useful in future studies to direct
high-level expression of substances for secretion into the lumen of the stomach to
test their importance for regulation of gastric physiology.
CONCLUSIONS
Integrative genomics, the study of gene function in genetically engineered animal models, takes advantage of the genome sequencing effort and the powerful
technologies for manipulating the mouse genome. Genes for many of the key
regulators of the stomach have been identified and cloned, and many knockout
models have been produced that have allowed further investigation of the roles
of these regulatory factors in the function of the gastric acid secretory system.
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
396
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
Although the data obtained from many genetically engineered mouse models have
supported the findings from more classical pharmacological and physiological experiments, the acid secretory phenotypes of other models were unexpected, often
resulting in the need to re-examine the roles of certain regulators. In particular,
the finding that basal acid secretion was normal in H2 receptor-deficient mice was
unexpected given that H2 receptor antagonists are extremely effective blockers of
acid secretion in the mouse. This finding alone suggests that there might be another
histamine pathway in parietal cells, that cAMP signaling is not necessary, or that
gastrin and/or ACh alone are sufficient for normal acid secretion. It is clear that
combining pharmacological and physiological experiments with the generation of
new genetically engineered mouse mutants will be required to fully understand the
complex regulation of the gastric acid secretory system. In the future, promoters
that have been defined for cell-specific transgene expression in the gastric mucosa can be utilized to create transgenics with altered gastric secretory function.
For example, manipulation of the cell signaling pathways in parietal cells using
the mouse H+/K+-ATPase β-subunit promoter would be useful to help define the
critical components for activation of acid secretion in this cell.
ACKNOWLEDGMENTS
Thanks to Dr. Renu Jain, Cindy Brunkan, Gina Bane, and Karen Ong for helpful
comments on the manuscript. Research in the laboratory of LC Samuelson is
supported by the National Institutes of Health. KL Hinkle was supported by the
Cellular and Molecular Aspects of Systems and Integrative Biology Training Grant
and by the Organogenesis Training Grant.
The Annual Review of Physiology is online at http://physiol.annualreviews.org
LITERATURE CITED
1. Hersey SJ, Sachs G. 1995. Gastric acid secretion. Physiol. Rev. 75:155–89
2. Zeng N, Kang T, Lyu RM, Wong H, Wen
Y, et al. 1998. The pituitary adenylate cyclase activating polypeptide type 1 receptor (PAC1-R) is expressed on gastric ECL
cells: evidence by immunocytochemistry
and RT-PCR. Ann. NY Acad. Sci. 865:147–
56
3. Pisegna JR, Ohning GV, Athmann C, Zeng
N, Walsh JH, Sachs G. 2000. Role of
PACAP1 receptor in regulation of ECL
cells and gastric acid secretion by pituitary
adenylate cyclase activating peptide. Ann.
NY Acad. Sci. 921:233–41
4. Cabero JL, Grapengiesser E, Gylfe E, Li
ZQ, Mardh S. 1992. Effects of gastrin on
cytosolic free Ca2+ in individual, acid-secreting rat parietal cells. Biochem. Biophys.
Res. Commun. 183:1097–102
5. Hill SJ. 1991. Histamine receptors and
interactions between second messenger
transduction systems. Agents Actions
Suppl. 33:145–59
6. Chew CS, Brown MR. 1986. Release of
intracellular Ca2+ and elevation of inositol
trisphosphate by secretagogues in parietal
and chief cells isolated from rabbit gastric
mucosa. Biochim. Biophys. Acta 888:116–
25
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
ACID SECRETION IN MOUSE MUTANTS
7. Li ZQ, Mardh S. 1996. Interactions between Ca2+- and cAMP-dependent stimulatory pathways in parietal cells. Biochim.
Biophys. Acta 1311:133–42
8. Urushidani T, Forte JG. 1997. Signal transduction and activation of acid secretion in
the parietal cell. J. Membr. Biol. 159:99–
111
9. Sandvik AK, Waldum HL. 1988. The effect of somatostatin on baseline and stimulated acid secretion and vascular histamine
release from the totally isolated vascularly
perfused rat stomach. Regul. Pept. 20:233–
39
10. Lucey MR, Yamada T. 1989. Biochemistry
and physiology of gastrointestinal somatostatin. Dig. Dis. Sci. 34:5S–13S (Suppl.)
11. Prinz C, Kajimura M, Scott DR, Mercier
F, Helander HF, Sachs G. 1993. Histamine
secretion from rat enterochromaffin-like
cells. Gastroenterology 105:449–61
12. Prinz C, Sachs G, Walsh JH, Coy DH,
Wu SV. 1994. The somatostatin receptor
subtype on rat enterochromaffin-like cells.
Gastroenterology 107:1067–74
13. Park J, Chiba T, Yamada T. 1987. Mechanisms for direct inhibition of canine gastric parietal cells by somatostatin. J. Biol.
Chem. 262:14190–96
14. Wyatt MA, Jarvie E, Feniuk W, Humphrey
PP. 1996. Somatostatin sst2 receptor-mediated inhibition of parietal cell function in rat
isolated gastric mucosa. Br. J. Pharmacol.
119:905–10
15. Zaki M, Harrington L, McCuen R, Coy DH,
Arimura A, Schubert ML. 1996. Somatostatin receptor subtype 2 mediates inhibition of gastrin and histamine secretion from
human, dog, and rat antrum. Gastroenterology 111:919–24
16. Rossowski WJ, Gu ZF, Akarca US, Jensen
RT, Coy DH. 1994. Characterization of somatostatin receptor subtypes controlling rat
gastric acid and pancreatic amylase release.
Peptides 15:1421–24
17. Lloyd KC, Wang J, Aurang K, Gronhed P,
Coy DH, Walsh JH. 1995. Activation of somatostatin receptor subtype 2 inhibits acid
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
397
secretion in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 268:G102–G6
Aurang K, Wang J, Lloyd KC. 1997. Somatostatin inhibition of acid and histamine
release by activation of somatostatin receptor subtype 2 receptors in rats. J. Pharmacol. Exp. Ther. 281:245–52
Fung LC, Greenberg GR. 1997. Characterization of somatostatin receptor subtypes
mediating inhibition of nutrient-stimulated
gastric acid and gastrin in dogs. Regul. Pept.
68:197–203
Lloyd KC, Amirmoazzami S, Friedik F,
Chew P, Walsh JH. 1997. Somatostatin inhibits gastrin release and acid secretion by
activating sst2 in dogs. Am. J. Physiol. Gastrointest. Liver Physiol. 272:G1481–G88
Karam S, Leblond CP. 1995. Origin and
migratory pathways of the eleven epithelial
cell types present in the body of the mouse
stomach. Microsc. Res. Tech. 31:193–214
Forte TM, Machen TE, Forte JG. 1977. Ultrastructural changes in oxyntic cells associated with secretory function: a membrane recycling hypothesis. Gastroenterology 73:941–55
Okamoto CT, Forte JG. 2001. Vesicular
trafficking machinery, the actin cytoskeleton, and H+-K+-ATPase recycling in the
gastric parietal cell. J. Physiol. 532:287–96
Koh TJ, Goldenring JR, Ito S, Mashimo H,
Kopin AS, et al. 1997. Gastrin deficiency
results in altered gastric differentiation and
decreased colonic proliferation in mice.
Gastroenterology 113:1015–25
Friis-Hansen L, Sundler F, Li Y, Gillespie
PJ, Saunders TL, et al. 1998. Impaired gastric acid secretion in gastrin-deficient mice.
Am. J. Physiol. Gastrointest. Liver Physiol.
274:G561–G68
Nagata A, Ito M, Iwata N, Kuno J, Takano
H, et al. 1996. G protein-coupled cholecystokinin-B/gastrin receptors are responsible for physiological cell growth of the
stomach mucosa in vivo. Proc. Natl. Acad.
Sci. USA 93:11825–30
Langhans N, Rindi G, Chiu M, Rehfeld JF,
Ardman B, et al. 1997. Abnormal gastric
14 Jan 2003
14:2
398
28.
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
29.
30.
31.
32.
33.
34.
35.
36.
37.
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
histology and decreased acid production
in cholecystokinin-B/gastrin receptor-deficient mice. Gastroenterology 112:280–86
Dockray GJ, Varro A, Dimaline R, Wang
T. 2001. The gastrins: their production
and biologial activities. Annu. Rev. Physiol. 63:119–39
Lloyd KC, Raybould HE, Tache Y, Walsh
JH. 1992. Role of gastrin, histamine, and
acetylcholine in the gastric phase of acid
secretion in anesthetized rats. Am. J. Physiol. Gastrointest. Liver Physiol. 262:G747–
G55
Ding XQ, Hakanson R. 1996. Effect of
cholecystokinin-B/gastrin receptor blockade on gastric acid secretion in conscious
rats. Pharmacol. Toxicol. 79:324–30
Walsh JH. 1990. Role of gastrin as a trophic
hormone. Digestion 47:11–16 (Suppl. 1)
Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, et al. 1996. Processing and proliferative effects of human progastrin in transgenic mice. J. Clin. Invest. 98:1918–29
Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, et al. 2000. Synergistic
interaction between hypergastrinemia and
Helicobacter infection in a mouse model of
gastric cancer. Gastroenterology 118:36–
47
Konda Y, Kamimura H, Yokota H, Hayashi
N, Sugano T, Takeuchi T. 1999. Gastrin
stimulates the growth of gastric pit with
less-differentiated features. Am. J. Physiol. Gastrointest. Liver Physiol. 277:G773–
G84
Scarff KL, Judd LM, Toh BH, Gleeson PA,
Van Driel IR. 1999. Gastrin H(+),K(+)adenosine triphosphatase beta subunit is required for normal function, development,
and membrane structure of mouse parietal
cells. Gastroenterology 117:605–18
Black JW, Duncan WA, Durant CJ,
Ganellin CR, Parsons EM. 1972. Definition
and antagonism of histamine H2 receptors.
Nature 236:385–90
Black J. 1993. Reflections on the analytical
pharmacology of histamine H2-receptor
antagonists. Gastroenterology 105:963–68
38. Kobayashi T, Tonai S, Ishihara Y, Koga R,
Okabe S, Watanabe T. 2000. Abnormal
functional and morphological regulation of
the gastric mucosa in histamine H2 receptor-deficient mice. J. Clin. Invest. 105:
1741–49
39. Tanaka S, Hamada K, Yamada N, Sugita
Y, Tonai S, et al. 2002. Gastric acid secretion in L-histidine decarboxylase-deficient
mice. Gastroenterology 122:145–55
40. Okabe S, Maeda K, Ogawa T, Kataoka
T, Furutani K, et al. 2002. Comparative
studies of gastric mucosal changes in histamine H2R-KO and histidine decarboxylase (HDC) KO mice. Gastroenterology
122(4):A-252 (Abstr.)
41. Vuyyuru L, Schubert ML. 1997. Histamine,
acting via H3 receptors, inhibits somatostatin and stimulates acid secretion in isolated mouse stomach. Gastroenterology
113:1545–52
42. Wank SA. 1995. Cholecystokinin receptors. Am. J. Physiol. Gastrointest. Liver
Physiol. 269:G628–G46
43. Aihara T, Fujishita T, Kataoka N, Kanatani
K, Amagase K, et al. 2002. Role of
muscarinic acetylcholine M3- and M1receptors in gastric acid secretion in mice;
studies with receptor knockout mice. Gastroenterology 122(4):A-252 (Abstr.)
44. Zheng H, Bailey A, Jiang MH, Honda K,
Chen HY, et al. 1997. Somatostatin receptor subtype 2 knockout mice are refractory to growth hormone-negative feedback on arcuate neurons. Mol. Endocrinol.
11:1709–17
45. Martinez V, Curi AP, Torkian B, Schaeffer
JM, Wilkinson HA, et al. 1998. High basal
gastric acid secretion in somatostatin receptor subtype 2 knockout mice. Gastroenterology 114:1125–32
46. Yang H, Wong H, Wu V, Walsh JH, Tache
Y. 1990. Somatostatin monoclonal antibody immunoneutralization increases gastrin and gastric acid secretion in urethaneanesthetized rats. Gastroenterology 99:
659–65
47. Varga G, Kisfalvi I Jr, Kordas K, Wong H,
14 Jan 2003
14:2
AR
AR177-PH65-16.tex
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
ACID SECRETION IN MOUSE MUTANTS
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
48.
49.
50.
51.
52.
53.
54.
55.
Walsh JH, Solomon TE. 1997. Effect of somatostatin immunoneutralization on gastric acid and pancreatic enzyme secretion
in anesthetized rats. J. Physiol. 91:223–27
Low MJ, Otero-Corchon V, Parlow AF,
Ramirez JL, Kumar U, et al. 2001. Somatostatin is required for masculinization
of growth hormone-regulated hepatic gene
expression but not of somatic growth. J.
Clin. Invest. 107:1571–80
Zeyda T, Diehl N, Paylor R, Brennan MB,
Hochgeschwender U. 2001. Impairment in
motor learning of somatostatin null mutant
mice. Brain Res. 906:107–14
Spicer Z, Miller ML, Andringa A, Riddle TM, Duffy JJ, et al. 2000. Stomachs
of mice lacking the gastric H,K-ATPase
alpha-subunit have achlorhydria, abnormal
parietal cells, and ciliated metaplasia. J.
Biol. Chem. 275:21555–65
Shull GE, Miller ML, Schultheis PJ. 2000.
Lessons from genetically engineered animal models VIII. Absorption and secretion of ions in the gastrointestinal tract.
Am. J. Physiol. Gastrointest. Liver Physiol.
278:G185–G90
Franic TV, Judd LM, Robinson D, Barrett
SP, Scarff KL, et al. 2001. Regulation of
gastric epithelial cell development revealed
in H(+)/K(+)-ATPase beta-subunit- and
gastrin-deficient mice. Am. J. Physiol.
Gastrointest. Liver Physiol. 281:G1502–
G11
Schultheis PJ, Clarke LL, Meneton P, Harline M, Boivin GP, et al. 1998. Targeted
disruption of the murine Na+/H+ exchanger
isoform 2 gene causes reduced viability of
gastric parietal cells and loss of net acid
secretion. J. Clin. Invest. 101:1243–53
Boivin GP, Schultheis PJ, Shull GE, Stemmermann GN. 2000. Variant form of diffuse
corporal gastritis in NHE2 knockout mice.
Comp. Med. 50:511–15
Lee MP, Ravenel JD, Hu RJ, Lustig LR,
Tomaselli G, et al. 2000. Targeted disruption of the Kvlqt1 gene causes deafness and
gastric hyperplasia in mice. J. Clin. Invest.
106:1447–55
399
56. Courtois-Coutry N, Roush D, Rajendran
V, McCarthy JB, Geibel J, et al. 1997. A
tyrosine-based signal targets H/K-ATPase
to a regulated compartment and is required
for the cessation of gastric acid secretion.
Cell 90:501–10
57. Wang T, Courtois-Coutry N, Giebisch G,
Caplan MJ. 1998. A tyrosine-based signal regulates H-K-ATPase-mediated potassium reabsorption in the kidney. Am. J.
Physiol. Renal Physiol. 275:F818–F26
58. Lorenz RG, Gordon JI. 1993. Use of transgenic mice to study regulation of gene expression in the parietal cell lineage of gastric units. J. Biol. Chem. 268:26559–70
59. Li Q, Karam SM, Gordon JI. 1995. Simian
virus 40 T antigen-induced amplification
of pre-parietal cells in transgenic mice. J.
Biol. Chem. 270:15777–88
60. Li Q, Karam SM, Gordon JI. 1996. Diphtheria toxin-mediated ablation of parietal
cells in the stomach of transgenic mice. J.
Biol. Chem. 271:3671–76
61. Canfield V, West AB, Goldenring JR, Levenson R. 1996. Genetic ablation of parietal
cells in transgenic mice: a new model for
analyzing cell lineage relationships in the
gastric mucosa. Proc. Natl. Acad. Sci. USA
93:2431–35
62. Karam SM, Li Q, Gordon JI. 1997. Gastric epithelial morphogenesis in normal and
transgenic mice. Am. J. Physiol. Gastrointest. Liver Physiol. 272:G1209–G20
63. Hocker M, Raychowdhury R, Plath T, Wu
H, O’Connor DT, et al. 1998. Sp1 and
CREB mediate gastrin-dependent regulation of chromogranin A promoter activity
in gastric carcinoma cells. J. Biol. Chem.
273:34000–7
64. Watkinson A, Dockray GJ. 1992. Functional control of chromogranin A and B
concentrations in the body of the rat stomach. Regul. Pept. 40:51–61
65. Dimaline R, Evans D, Forster ER, Sandvik
AK, Dockray GJ. 1993. Control of gastric
corpus chromogranin A messenger RNA
abundance in the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 264:G583–88
14 Jan 2003
14:2
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
400
AR
AR177-PH65-16.tex
SAMUELSON
¥
AR177-PH65-16.sgm
LaTeX2e(2002/01/18)
P1: IBC
HINKLE
66. Syversen U, Mignon M, Bonfils S, Kristensen A, Waldum HL. 1993. Chromogranin A and pancreastatin-like immunoreactivity in serum of gastrinoma patients. Acta
Oncol. 32:161–65
67. Borch K, Stridsberg M, Burman P, Rehfeld
JF. 1997. Basal chromogranin A and gastrin concentrations in circulation correlate
to endocrine cell proliferation in type-A
gastritis. Scand. J. Gastroenterol. 32:198–
202
68. Hocker M, Cramer T, O’Connor DT, Rosewicz S, Wiedenmann B, et al. 2001. Neuroendocrine-specific and gastrin-dependent
expression of a chromogranin A-luciferase
fusion gene in transgenic mice. Gastroenterology 121:43–55
69. Zhukova E, Afshar A, Ko J, Popper P, Pham
T, et al. 2001. Expression of the human insulin gene in the gastric G cells of transgenic mice. Transgenic Res. 10:329–41
70. Winston JH, Hanten GR, Overbeek PA,
Kellems RE. 1992. 50 flanking sequences
of the murine adenosine deaminase gene
direct expression of a reporter gene to specific prenatal and postnatal tissues in transgenic mice. J. Biol. Chem. 267:13472–79
71. Xu PA, Winston JH, Datta SK, Kellems RE.
1999. Regulation of forestomach-specific
expression of the murine adenosine deaminase gene. J. Biol. Chem. 274:10316–23
72. Chinsky JM, Ramamurthy V, Fanslow WC,
Ingolia DE, Blackburn MR, et al. 1990.
Developmental expression of adenosine
deaminase in the upper alimentary tract of
mice. Differentiation 42:172–83
P1: FDS
January 17, 2003
11:23
Annual Reviews
AR177-FM
Annual Review of Physiology,
Volume 65, 2003
CONTENTS
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
Frontispiece—Jean D. Wilson
xiv
PERSPECTIVES, Joseph F. Hoffman, Editor
A Double Life: Academic Physician and Androgen Physiologist,
Jean D. Wilson
1
CARDIOVASCULAR PHYSIOLOGY, Jeffrey Robbins, Section Editor
Lipid Receptors in Cardiovascular Development, Nick Osborne
and Didier Y.R. Stainier
Cardiac Hypertrophy: The Good, the Bad, and the Ugly, N. Frey
and E.N. Olson
Stress-Activated Cytokines and the Heart: From Adaptation to
Maladaptation, Douglas L. Mann
23
45
81
CELL PHYSIOLOGY, Paul De Weer, Section Editor
Cell Biology of Acid Secretion by the Parietal Cell, Xuebiao Yao
and John G. Forte
Permeation and Selectivity in Calcium Channels, William A. Sather
and Edwin W. McCleskey
Processive and Nonprocessive Models of Kinesin Movement,
Sharyn A. Endow and Douglas S. Barker
103
133
161
COMPARATIVE PHYSIOLOGY, George N. Somero, Section Editor
Origin and Consequences of Mitochondrial Variation in Vertebrate
Muscle, Christopher D. Moyes and David A. Hood
Functional Genomics and the Comparative Physiology of Hypoxia,
Frank L. Powell
Application of Microarray Technology in Environmental
and Comparative Physiology, Andrew Y. Gracey and
Andrew R. Cossins
177
203
231
ENDOCRINOLOGY, Bert W. O’Malley, Section Editor
Nuclear Receptors and the Control of Metabolism,
Gordon A. Francis, Elisabeth Fayard, Frédéric Picard, and
Johan Auwerx
261
vii
P1: FDS
January 17, 2003
viii
11:23
Annual Reviews
AR177-FM
CONTENTS
Insulin Receptor Knockout Mice, Tadahiro Kitamura, C. Ronald Kahn,
and Domenico Accili
The Physiology of Cellular Liporegulation, Roger H. Unger
313
333
GASTROINTESTINAL PHYSIOLOGY, John Williams, Section Editor
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
The Gastric Biology of Helicobacter pylori, George Sachs,
David L. Weeks, Klaus Melchers, and David R. Scott
Physiology of Gastric Enterochromaffin-Like Cells, Christian Prinz,
Robert Zanner, and Manfred Gratzl
Insights into the Regulation of Gastric Acid Secretion Through Analysis of
Genetically Engineered Mice, Linda C. Samuelson and Karen L. Hinkle
349
371
383
NEUROPHYSIOLOGY, Richard Aldrich, Section Editor
In Vivo NMR Studies of the Glutamate Neurotransmitter Flux
and Neuroenergetics: Implications for Brain Function,
Douglas L. Rothman, Kevin L. Behar, Fahmeed Hyder,
and Robert G. Shulman
401
Transducing Touch in Caenorhabditis elegans, Miriam B. Goodman
and Erich M. Schwarz
429
Hyperpolarization-Activated Cation Currents: From Molecules
to Physiological Function, Richard B. Robinson and
Steven A. Siegelbaum
453
RENAL AND ELECTROLYTE PHYSIOLOGY, Steven C. Hebert, Section Editor
Macula Densa Cell Signaling, P. Darwin Bell, Jean Yves Lapointe,
and János Peti-Peterdi
Paracrine Factors in Tubuloglomerular Feedback: Adenosine, ATP,
and Nitric Oxide, Jürgen Schnermann and David Z. Levine
Regulation of Na/Pi Transporter in the Proximal Tubule,
Heini Murer, Nati Hernando, Ian Forster, and Jürg Biber
Mammalian Urea Transporters, Jeff M. Sands
Terminal Differentiation of Intercalated Cells: The Role of Hensin,
Qais Al-Awqati
481
501
531
543
567
RESPIRATORY PHYSIOLOGY, Carole R. Mendelson, Section Editor
Current Status of Gene Therapy for Inherited Lung Diseases,
Ryan R. Driskell and John F. Engelhardt
The Role of Exogenous Surfactant in the Treatment of Acute Lung
Injury, James F. Lewis and Ruud Veldhuizen
Second Messenger Pathways in Pulmonary Host Defense,
Martha M. Monick and Gary W. Hunninghake
585
613
643
P1: FDS
January 17, 2003
11:23
Annual Reviews
AR177-FM
CONTENTS
Alveolar Type I Cells: Molecular Phenotype and Development,
Mary C. Williams
Annu. Rev. Physiol. 2003.65:383-400. Downloaded from arjournals.annualreviews.org
by University of Malaga on 08/15/06. For personal use only.
SPECIAL TOPIC: LIPID RECEPTOR PROCESSES, Donald W. Hilgemann,
Special Topic Editor
Getting Ready for the Decade of the Lipids, Donald W. Hilgemann
Aminophospholipid Asymmetry: A Matter of Life and Death,
Krishnakumar Balasubramanian and Alan J. Schroit
Regulation of TRP Channels Via Lipid Second Messengers,
Roger C. Hardie
Phosphoinositide Regulation of the Actin Cytoskeleton,
Helen L. Yin and Paul A. Janmey
Dynamics of Phosphoinositides in Membrane Retrieval and
Insertion, Michael P. Czech
SPECIAL TOPIC: MEMBRANE PROTEIN STRUCTURE, H. Ronald Kaback,
Special Topic Editor
Structure and Mechanism of Na,K-ATPase: Functional Sites
and Their Interactions, Peter L. Jorgensen, Kjell O. Håkansson,
and Steven J. Karlish
G Protein-Coupled Receptor Rhodopsin: A Prospectus,
Slawomir Filipek, Ronald E. Stenkamp, David C. Teller, and
Krzysztof Palczewski
ix
669
697
701
735
761
791
817
851
INDEXES
Subject Index
Cumulative Index of Contributing Authors, Volumes 61–65
Cumulative Index of Chapter Titles, Volumes 61–65
ERRATA
An online log of corrections to Annual Review of Physiology chapters
may be found at http://physiol.annualreviews.org/errata.shtml
881
921
925