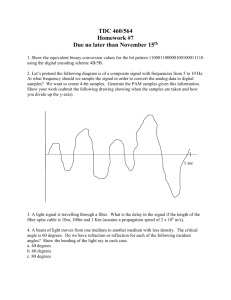
Fiber Spinning and Characterization
advertisement

2016/3/7 Introduction to Fiber Spinning Characterization Qing Shen Donghua University, Shanghai, China and 2 Preface Contents 1. Introduction 1.1 Fiber history 1.2 Fiber classification 1.3 Importance of fiber science and technology Recommending reading 2. Methods for fiber spinning 2.1 Melt spinning 2.2 Wet spinning 2.3 Solution dry spinning 2.4 Solution wet spinning 2.5 Gel spinning 2.6 Liquid crystal spinning 2.7 Electrospinning 2.8 Bi-components spinning 2.9 Reaction spinning 2.10 Centrifugation spinning 2.11 Emulsion spinning 2.12 Interfacial polycondensation spinning 2.13 Laser associated spinning 2.14 Coaxial spinning 2.15 Dry spinning 2.16 Dry jet-wet spinning 2.17 Polymerization method Recommending reading 3. Methods for fiber characterization 3.1 Morphology 3.1.1 Optical microscopy 3.1.2 SEM 3.1.3 TEM 3.1.4 AFM 3.1.5 STM 3.2 Thermal properties 3.2.1 DSC 3.2.2 TG/DTG 3.2.3 DMA 3.3 Mechanical properties 3.3.1 Tensile 3.3.2 Elongation 3.3.3 Modulus 2 3 3.4 Structure 3.4.1 XRD 3.4.2 FTIR 3.4.3 FT-Raman 3.4.4 NMR 3.4.5 UV 3.5 Bio-properties 3.6 Nano-properties Recommending reading 4. Typical materials and related fiber spinning techniques 4.1 Synthetic polymer fibers 4.1.1 PA 4.1.2 PAN 4.1.3 PP 4.1.4 PET/PBT/PTT 4.1.5 PU 4.1.6 PVA 4.1.7 PVC 4.1.8 PTFE 4.1.9 PLA 4.2 Natural polymer fibers 4.2.1 Cellulose 4.2.2 Chitin 4.2.3 Protein 4.2.4 Lignin 4.3 Inorganic fibers 4.3.1 Carbon fiber 4.3.2 Silicon fiber 4.3.3 Glass fiber 4.3.4 Oxide fiber Recommending reading 3 4 1. Introduction 1.1 Fiber development history Fiber is defined as a solid material with stable thin shape and long size as well as certain level of tensile strength. As has been consistently recognized that the fiber science and technology were learnt from silkworm and spider since their fiber producing processes are good examples of the biosynthetic and biospinning techniques because they convert non-fiber foods by enzymes into proteins in body then spun fibers as a cocoon or net. In addition to animal fibers, plants-based fibers are generally synthesized in natural conditions, e.g. by photo, carbon dioxide and water. The use of fiber in human history can back to the Paleolithic times by observing ancient people used cordage in fishing, trapping, and transport, and in fabrics for clothing using palm leaf, jute, flax, ramie, sedges, rushes, and reeds as raw materials. The first true paper is believed to have been made in southeastern China in the second century AD from old rags, bast fibers of hemp and ramie and later from the bast fiber of the mulberry tree. For thousands of years, the use of fiber was limited by the inherent qualities available in the natural world. Cotton and linen wrinkled from wear and washings. Silk required delicate handling. Wool shrank, was irritating to the touch, and was eaten by moths. Then, a mere century ago, rayon —the first manufactured fiber—was developed. The secrets of fiber chemistry for countless applications had begun to emerge. Manufactured fibers now are put to work in modern apparel, home furnishings, medicine, aeronautics, energy, industry, and more. Fiber engineers can combine, modify and tailor fibers in ways far beyond the performance limits of fiber drawn from the silkworm cocoon, grown in the fields, or spun from the fleece of animals. The Early Attempts The earliest published record of an attempt to create an artificial fiber took place in 1664. English naturalist Robert Hooke suggested the possibility of producing a fiber that would be “if not fully as good, nay better” than silk. His goal remained unachieved for more than two centuries. The first patent for “artificial silk” was granted in England in 1855 to a Swiss chemist named Audemars. He dissolved the fibrous inner bark of a mulberry tree, chemically modifying it to produce cellulose. He formed threads by dipping needles into this solution and drawing them out but it never occurred to him to emulate the silkworm by extruding the cellulosic liquid through a small hole. In the early 1880's, Sir Joseph W. Swan, an English chemist and electrician, was spurred to action by Thomas Edison's new incandescent electric lamp. He experimented with forcing a liquid similar to Audemars solution through fine holes into a coagulating bath. His fibers worked like carbon filament, and they found early use in Edison's invention. It also occurred to Swan that his filament could be used to make textiles. In 1885 he exhibited in London some fabrics crocheted by his wife from his new fiber. But electrical lamps remained his main interest, and he soon abandoned work on textile applications. First Commercial Production 4 5 The first commercial scale production of a manufactured fiber was achieved by French chemist Count Hilaire de Chardonnet. In 1889, his fabrics of “artificial silk” caused a sensation at the Paris Exhibition. Two years later he built the first commercial rayon plant at Besancon, France, and secured his fame as the “father of the rayon industry.” Several attempts to produce “artificial silk” in the United States were made during the early 1900's but none were commercially successful until the American Viscose Company, formed by Samuel Courtaulds and Co., Ltd., began production its production of rayon in 1910. In 1893, Arthur D. Little of Boston, invented yet another cellulosic product — acetate — and developed it as a film. By 1910, Camille and Henry Dreyfus were making acetate motion picture film and toilet articles in Basel, Switzerland. During World War I, they built a plant in England to produce cellulose acetate dope for airplane wings and other commercial products. Upon entering the War, the United States government invited the Dreyfus brothers to build a plant in Maryland to make the product for American warplanes. The first commercial textile uses for acetate in fiber form were developed by the Celanese Company in 1924. In the meantime, U.S. rayon production was growing to meet increasing demand. By the mid-1920's, textile manufacturers could purchase the fiber for half the price of raw silk. So began manufactured fibers' gradual conquest of the American fiber market. This modest start in the 1920's grew to nearly 70% of the national market for fiber by the last decade of the century. Nylon — The “Miracle” Fiber In September 1931, American chemist Wallace Carothers reported on research carried out in the laboratories of the DuPont Company on “giant” molecules called polymers. He focused his work on a fiber referred to simply as “66,” a number derived from its molecular structure. Nylon, the “miracle fiber,” was born. The Chemical Heritage Foundation is currently featuring an exhibit on the history of nylon. By 1938, Paul Schlack of the I.G. Farben Company in Germany, polymerized caprolactam and created a different form of the polymer, identified simply as nylon “6.” Nylon's advent created a revolution in the fiber industry. Rayon and acetate had been derived from plant cellulose, but nylon was synthesized completely from petrochemicals. It established the basis for the ensuing discovery of an entire new world of manufactured fibers. An American Romance DuPont began commercial production of nylon in 1939. The first experimental testing used nylon as sewing thread, in parachute fabric, and in women's hosiery. Nylon stockings were shown in February 1939 at the San Francisco Exposition — and the most exciting fashion innovation of the age was underway. 5 6 American women had only a sampling of the beauty and durability of their first pairs of nylon hose when their romance with the new fabric was cut short. The United States entered World War II in December 1941 and the War Production Board allocated all production of nylon for military use. Nylon hose, which sold for $ 1.25 a pair before the War, moved in the black market at $10. Wartime pin-ups and movie stars, like Betty Grable, auctioned nylon hose for as much as $40,000 a pair in war-effort drives. During the War, nylon replaced Asian silk in parachutes. It also found use in tires, tents, ropes, ponchos, and other military supplies, and even was used in the production of a high-grade paper for U.S. currency. At the outset of the War, cotton was king of fibers, accounting for more than 80% of all fibers used. Manufactured and wool fibers shared the remaining 20%. By the end of the War in August 1945, cotton stood at 75% of the fiber market. Manufactured fibers had risen to 15%. The Post-War Industry After the war, GI's came home, families were reunited, industrial America gathered its peacetime forces, and economic growth surged. The conversion of nylon production to civilian uses started and when the first small quantities of postwar nylon stockings were advertised, thousands of frenzied women lined up at New York department stores to buy. In the immediate post-war period, most nylon production was used to satisfy this enormous pent up demand for hosiery. But by the end of the 1940's, it was also being used in carpeting and automobile upholstery. At the same time, three new generic manufactured fibers started production. Dow Badische Company (today, BASF Corporation) introduced metalized fibers; Union Carbide Corporation developed modacrylic fiber; and Hercules, Inc. added olefin fiber. Manufactured fibers continued their steady march. By the 1950's, the industry was supplying more than 20% of the fiber needs of textile mills. A new fiber, “acrylic,” was added to the list of generic names, as DuPont began production of this wool-like product. Meanwhile, polyester, first examined as part of the Wallace Carothers early research, was attracting new interest at the Calico Printers Association in Great Britain. There, J. T. Dickson and J. R. Whinfield produced a polyester fiber by condensation polymerization of ethylene glycol with terephthalic acid. DuPont subsequently acquired the patent rights for the United States and Imperial Chemical Industries for the rest of the world. A host of other producers soon joined in. A Wash and Wear Revolution In the summer of 1952, “wash and wear” was coined to describe a new blend of cotton and acrylic. The term eventually was applied to a wide variety of manufactured fiber blends. Commercial production of polyester fiber transformed the “wash and wear” novelty into a revolution in textile product performance. 6 7 Polyester's commercialization in 1953 was accompanied by the introduction of triacetate. The majority of the 20th century's basic manufactured fibers now had been discovered, and the industry's engineers turned to refining their chemical and physical properties to extend their use across the American economy. In the 1960's and 1970's consumers bought more and more clothing made with polyester. Clotheslines were replaced by electric dryers, and the “wash and wear” garments they dried emerged wrinkle free. Ironing began to shrink away on the daily list of household chores. Fabrics became more durable and color more permanent. New dyeing effects were being achieved and shape-retaining knits offered new comfort and style. Endless Possibilities In the 1960's, manufactured fiber production accelerated as it was spurred on by continuous fiber innovation. The revolutionary new fibers were modified to offer greater comfort, provide flame resistance, reduce clinging, release soil, achieve greater whiteness, special dullness or luster, easier dyeability, and better blending qualities. New fiber shapes and thicknesses were introduced to meet special needs. Spandex, a stretchable fiber; aramid, a high-temperature-resistant polyamide; and para-aramid, with outstanding strength-to-weight properties, were introduced into the marketplace. In the early 1960's, manufactured fiber accounted for nearly 30% of American textile mill consumption. By 1965, the manufactured fiber industry was providing over 40% of the nation's fiber needs. One dramatic new set of uses for manufactured fibers came with the establishment of the U.S. space program. The industry provided special fiber for uses ranging from clothing for the astronauts to spaceship nose cones. When Neil Armstrong took “One small step for man, one giant leap for mankind,” on the moon on July 20, 1969, his lunar space suit included multi-layers of nylon and aramid fabrics. The flag he planted was made of nylon. Today, the exhaust nozzles of the two large booster rockets that lift the space shuttle into orbit contain 30,000 pounds of carbonized rayon. Carbon fiber composites are used in as structural components in the latest commercial aircraft, adding strength and lowering weight and fuel costs. Safety and Energy Challenges The early 1970's saw a wave of consumer protection demands, most notably one for a mandated Federal flammability standard for children's sleepwear. The manufactured fiber industry spent $20 million on flammability research and development in 1972 and 1973, and manufactured fiber fabrics became predominant in this market. Flammability standards were also issued for carpet and other products. In the U.S. carpet market, 99% of all surface fibers are now manufactured fibers. 7 8 In late 1973, when the Nation was struck by a severe energy crisis, the manufactured fiber industry reduced the energy required to produce a pound of fiber by 26%. By then, the industry was using but 1% of the Nation's petroleum supply to provide two-thirds of all fibers used by American textile mills. Today Innovation is the hallmark of the manufactured fiber industry. Fibers more numerous and diverse than any found in nature are now routinely created in the industry's laboratories. Nylon variants, polyester, and olefin are used to produce carpets that easily can be rinsed clean — even 24 hours after they've been stained. Stretchable spandex and machine-washable, silk-like polyesters occupy solid places in the U.S. apparel market. The finest microfibers are remaking the world of fashion. For industrial uses, manufactured fibers relentlessly replace traditional materials in applications from super-absorbent diapers, to artificial organs, to construction materials for moon-based space stations. Engineered non-woven products of manufactured fibers are found in applications from surgical gowns and apparel interfacing to roofing materials, road bed stabilizers, and floppy disk envelopes and liners. Non-woven fabrics, stiff as paper or as soft and comfortable as limp cloth, are made without knitting or weaving. As they always have, manufactured fibers continue to mean, “life made better.” First Commercial U.S. Production 1910 — Rayon 1941 — Saran 1959 — Spandex 1924 — Acetate 1946 — Metallic 1961 —Aramid 1930 — Rubber 1949 — Modacylic 1983 — PBI 1936 — Glass 1949 — Olefin 1983 — Sulfar 1939 — Nylon 1950 — Acrylic 1992 — Lyocell 1939 — Vinyon 1953 — Polyester The first chemical fiber was cellulose-based produced by Nicolaus de Chardonnet in 1884. After that in 1898 in Oberbruch near Aachen of Germany, Paul Fremery, Bromert and Urban produced the first cooper silk filaments. Polyvinyl chloride, PVC, was the first fully synthetic fiber that was produced by Klatte (1913). Then, in 1927 Staudinger succeeded spun the fully synthetic fiber from polyoxymethylene and later from polyethylenoxide from the melt. In 1934, the first semitechnical production of polyaerylonitrile fibers (PAN) was reported in Germany. The polyurethane, PU, fiber was developed in 1937 by O. Bayer et al. In 1938, Carothers reported the first polycondensation fiber, Nylon, in the company DuPont de Nemours & Co. Following, 8 9 Schlack produced lactam-based fiber, Perlon®, in 1939 in Berlin-Lichtenberg, Germany. The commercial polyester fibers were reported around 1950 developed by Whinefield, and polypropylene, PP, around 1958 developed by NaUa. The high performance fiber, e.g. Nomex® and Kevlar® both were developed by DuPont in 1963 and 1970, respectively, will be mentioned here. The development of high-grade carbon fibers was in 1966 with the oxidation and carbonization of PAN filaments. Many of the developed fibers have been observed without commercialized, e.g., polyaminotriazoles fiber, polyamides 4 and 7, PA 4 and PA 7, due to spinning difficult or cost reasons. The novel biodegradable fiber, poly(lactic acid), PLA, was developed by Cargill Dow Chemical company in 1997. 1.2 Fiber structure The fiber structure is generally known in micro and macro as Figures 1 and 2 described, respectively. Figure 1. A scheme of the micro structure of fiber. Basically, it was known that the fiber consists of three distinct phases, i.e. the oriented crystalline regions, the amorphous regions also with preferential orientation along the fiber axis which contain tie molecules connecting crystallites, and the highly extended non-crystalline molecules which was called the interfibrillar phase. In these three phases, the interfibrillar phase plays a key role in the tensile properties of the fiber. 9 10 Figure 2. A scheme of the macro structure of fiber.[] 1.3 Fiber classification The classification of fiber has been difined by many peoples and a diameter-based scale to be as Figure 3 described. The classification of man-made fibres Man-made fibres are classified into three classes, those made from natural polymers, those made from synthetic polymers and those made from inorganic materials. Fibres from Natural Polymers The most common natural polymer fibre is viscose, which is made from the polymer cellulose obtained mostly from farmed trees. Other cellulose-based fibres are cupro, acetate and triacetate, lyocell and modal. The production processes for these fibres are given in Part 4. Less common natural polymer fibres are made from 10 11 rubber, alginic acid and regenerated protein. Fibres from Synthetic Polymers There are very many synthetic fibres i.e. organic fibres based on petrochemicals. The most common are polyester, polyamide (often called nylon), acrylic and modacrylic, polypropylene, the segmented polyurethanes which are elastic fibres known as elastanes (or spandex in the USA), and speciality fibres such as the high performance aramids. Fibres from Inorganic Materials The inorganic man-made fibres are fibres made from materials such as glass, metal, carbon or ceramic. These fibres are very often used to reinforce plastics to form composites. There are several fibres made from the naturally occurring polymer cellulose which is present in all plants. Mostly cellulose from wood is used to produce the fibres but sometimes cellulose from short cotton fibres, called linters, is the source. By far the most common cellulosic fibre is viscose. Viscose is defined very simply by BISFA as being "a cellulose fibre obtained by the viscose process". It is known as rayon in the USA. Although several cellulosic fibres had been made experimentally during the 19th century, it was not until 1905 that what has become the most popular cellulosic fibre, viscose, was produced. Production Viscose fibres are made from cellulose from wood pulp. The cellulose is ground up and reacted with caustic soda. After a waiting period, the ripening process during which depolymerisation occurs, carbon disulphide is added. This forms a yellow crumb known as cellulose xanthate, which is easily dissolved in more caustic soda to give a viscous yellow solution. This solution is pumped through a spinneret, which may contain thousands of holes, into a dilute sulphuric acid bath where the cellulose is regenerated as fine filaments as the xanthate decomposes. (See annex 1) Properties and End-Uses Viscose fibres, like cotton, have a high moisture regain. It dyes easily, it does not shrink when heated, and it is biodegradable. It is used in most apparel end-uses, often blended with other fibres, and in hygienic disposables where its high absorbency gives advantages. In filament yarn form it is excellent for linings. It is used very little in home furnishing fabrics but in the industrial field, because of its thermal stability, a high modulus version is still the main product used in Europe to reinforce high speed tyres. 11 12 Modal Fibres and polynosic fibres are both high wet modulus fibres made by the viscose process but with a higher degree of polymerisation and modified precipitating baths. This leads to fibres with improved properties such as better wear, higher dry and wet strengths and better dimensional stability. 12 13 Acetate and triacetate The term acetate fibres is used to describe fibres made from cellulose acetate. The difference between diacetate and triacetate fibres lies in the number of the cellulose hydroxyl groups that are acetylated. For acetate fibres the number lies between 75% and 92%, for triacetate fibres it is more than 92%. Production Wood cellulose is swollen by acetic acid and then converted to cellulose acetate using acetic anhydride and it is then dissolved in acetone. The resulting viscous solution is pumped through spinnerets into warm air to form filaments. The acetone evaporates and is recovered. The filaments are then wound up as filament yarns or collected as a tow. Properties and End-Uses These fibres are different from viscose in that they melt, are dyed using disperse dyes, absorb little water and can be textured. Although the dry strength of the two types are similar, triacetate has a higher wet strength. It also has a high melting point, 300 degrees C, compared with 250 degrees C for diacetate. Main end-uses for the filament yarns are linings and dresswear. There is very little staple fibre made from these fibres but acetate tow is the major product used for cigarette filters. Cupro cellulosic fibre was first produced commercially in Germany in 1908. Production Cotton cellulose is first bleached by boiling in an alkaline solution. This is then dissolved in a mixture of copper oxide and ammonia (the cuprammonium solvent). The blue viscous liquid is pumped through the spinneret into a spinning tube in which weak alkaline water is flowing. This water flow stretches the filaments before they are dried and wound up. Properties and End-Uses Cupro fibres have a good drape and are easy to wash. The main production is in filament yarn form for woven fabrics, largely for linings. 13 14 A new generation of cellulosic appeared in the market in December 1992 when a commercial plant in the USA started to make a lyocell staple fibre, based largely on European man-made fibre industry research. Subsequently, two European production plants have opened. Production The process used to make lyocell fibres is a solvent spinning process. The cellulose is dissolved in the solvent N-methylmorpholine n-oxide (NMMO) containing just the right amount of water. The solution is then filtered and spun through spinnerets to make the filaments, which are spun into water. The NMMO solvent is recovered from this aqueous solution and reused. Properties and End-Uses The lyocell fibres, like other cellulosics, are moisture absorbent and biodegradable. They have a dry strength higher than other cellulosics and approaching that of polyester. They also retain 85% of their strength when wet. Under certain conditions lyocell fibres fibrillate which enables fabrics to be developed with interesting aesthetics. Non- fibrillating versions are also available. Lyocell fibres are mostly used for apparel fabrics, especially outerwear, but it has been shown that, due to the fibrillating property some very interesting nonwoven fabrics can be made. Acrylic and Modacrylic BISFA defines acrylic fibres as "fibres composed of linear macromolecules having in the chain at least 85% (by mass) of acrylonitrile repeating units". Modacrylic fibres have, in the chain, at least 50% and less than 85% by mass of acrylonitrile. The first commercial fibres were introduced in the USA and Germany in 1948. Production The starting materials for acrylonitrile are propylene and ammonia, which are reacted with oxygen in the presence of catalysts. The acrylonitrile is then polymerised to produce polyacrylonitrile (PAN). The PAN is then spun into fibres from a solution in a solvent. Two process routes are used, wet spinning in which the fibres are spun into an aqueous coagulation bath and dry spinning in which the fibres are spun into hot air. (see annex 2) The fibres are then stretched, washed and crimped. The modacrylic fibres contain halogen comonomers such as vinyl chloride or vinylidene chloride, and have flame-retardant properties. (Chart 3) Properties and End-Uses Acrylic fibres are soft, flexible and have a high loft. For this reason they are widely used in knitted apparel end-uses such as sweaters and socks. (Chart 4) In addition to knitted apparel, home furnishing and blankets are other important applications. Acrylic fibres are used as a precursor for producing carbon fibre. 14 15 Polyamide A polyamide fibre is defined by BISFA as being "a fibre composed of linear macromolecules having in the chain recurring amide linkages, at least 85% of which are joined to aliphatic or cycloaliphatic units". There are many polyamide fibres made but only two, described below, are made in significant quantities. The first fibres made from polyamide polymers were produced in 15 16 1938 in the USA and Germany. In the USA the raw materials, which were used to produce the polymer, were adipic acid and hexamethylene diamine. Since both chemicals contain 6 carbon atoms the new polymer was named polyamide 6.6. In Germany caprolactam was polymerised to produce a different fibre known as polyamide 6. Production To produce fibres from the polyamide polymers the molten polymer is pumped through spinneret holes at a temperature approaching 300 degrees C to form filaments that are cooled and solidified in a quench air stream. If filament yarn is being produced the filaments are then oiled and wound onto cylinders. Polyamide yarns are spun to different orientations depending upon the use. If a fully oriented yarn (FOY or FDY) is required it is achieved by having a draw stage on the spinning machine, a process called spindrawing, or by spinning the yarn at very high speeds. (see annex 3) If the yarn is to be textured the preferred orientation is partial (POY). This yarn is then fully drawn and textured in a separate process. If staple fibres are being produced, very many filaments are bundled together to form a tow which is subsequently stretched, crimped and cut to the desired length. In 1999 there were over 3.4 million tonnes of polyamide filament produced worldwide and over 0.5 million tonnes of polyamide staple. (Chart 5) Properties and End-Uses In weaving the main end-use is for outerwear and technical fabrics. In knitting, stockings and tights and outerwear are both important outlets. Carpets and ropes and twines are also important sectors. (Chart 6) 16 17 A polyester, according to BISFA, is "a fibre composed of linear macromolecules having a chain at least 85% by mass of a diol and terephthalic acid". The first polyester was made in the UK in 1941. This polyester, known as polyethylene terephthalate(PET) has become the world's major man- made fibre. Other polyesters such as polybutylene terephthalate (PBT) and polytrimethylene terephthalate (PTT) are made but in much smaller 17 18 quantities. Production Polyester fibres are made in a very similar way to polyamide. Some plants take polyester polymer chips and melt them, at around 280 degrees C and then extrude the melt into continuous filaments to be wound onto packages or collected in cans as a tow before being stretched, crimped and cut into staple fibre. Other plants produce the polymer by a continuous process (CP) and form it into fibres without producing chips. A growing quantity is made by recycling PET bottles and other waste. If fully oriented yarns (FOY) are being produced the fibres are drawn on the spinning machine. If the yarn is to be textured, partially oriented yarns (POY) are spun. (see annex 4 - Chart 7) Properties and End-Uses In Western Europe, apparel accounts for a large share of usage of polyester fibres. Industrial use, such as tyre fabrics, and unspun uses such as furniture fillings and nonwovens, are both expanding rapidly. (Chart 8) 18 19 There are two polyolefin polymers used to make synthetic fibres, polypropylene and polyethylene, with polypropylene being by far the most important. The BISFA definition for polyethylene fibres is "fibre composed of linear macromolecules of unsubstituted saturated aliphatic hydrocarbons" and for polypropylene fibres "fibre composed of linear macromolecules made up of saturated aliphatic carbon units in which one carbon atom in 19 20 two carries a methyl side group...". Polyethylene was first produced in the UK in 1933 by polymerising ethylene under pressure. In 1938 in Germany polyethylene was made by polymerising ethylene in an emulsion. Polypropylene was commercialised in 1956 by polymerising propylene using catalysts. Both of these polyolefins are very important in plastic moulding and for making plastic sheet but both are spun into synthetic fibres on a large scale. Production Polyolefin fibres are made by melt spinning. Usually polymer granules - made by specialist producers rather than fibre companies - are fed to an extruder which melts the polymer which is then pumped through a spinneret. The filaments are cooled in an air stream before being wound on a package or collected in cans as a tow. Because the fibres are difficult to dye, coloured pigments are often added to the polymer stream before extrusion. An alternative process is to produce a film, cut the film into strips and then fibrillate the individual strips before winding onto a package. Recently a new family of catalysts to make polypropylene has been developed called metallocene catalysts. It is claimed that the polymers made from these catalysts can be spun to finer counts and drawn to give higher tenacities than existing polymers. (Chart 9) Properties and End-Uses Both polyolefin fibres have a density less than 1.0 and therefore, at a given decitex, are thicker than other man-made fibres and give more cover. They do not absorb moisture, which is an advantage in many end- uses, but without modification, they cannot be dyed. Their melting points are around 130 degrees C for polyethylene and 160 degrees C for polypropylene. They have a high resistance to chemical attack and modern polypropylene fibres have a high resistance to UV degradation. Polypropylene fibre consumption has grown rapidly during the past decade. This is due largely to its acceptance as a carpet fibre and the growth in the nonwoven end-uses, especially disposables and geotextiles where polypropylene is now the dominant fibre. (Chart 10) In 1998 370,000 tonnes of polypropylene were used in carpets, 228,000 tonnes in hygiene and medical and 106,000 tonnes in geotextiles and agrotextiles. In addition to these products the properties of the polyolefins make them ideal for end uses such as ropes, tapes, twines, fishing nets and sacking (from slit film). Speciality fibres A speciality fibre can be described as being a fibre with unique properties which make it the preferred fibre for particular applications. They are more expensive than the other synthetic fibres described in this document and are produced in comparatively small volumes. Statistics about the production and consumption of these fibres are not widely available. 20 21 There are very many speciality, or niche, fibres. Aramids Aramid is a contraction of aromatic and polyamide. BISFA defines these fibres as "fibre composed of linear macromolecules made up of aromatic groups joined by amide or imide linkages". There are two types of aramid: the meta-aramids and the paraaramids. The development of aramid fibres took place during the mid-50's to mid- 60's in the USA. The fibres were shown to have high melting points and high moduli. The impetus to develop the fibres came for the need for high performance fibres for air and space travel. Production The polymer polymphenyleneisophthalamide is used to make meta- aramids and the polymer polypterephthalamide to make paraaramids. Because the aramids decompose before they melt they are produced by wet and dry spinning methods. Sulphuric acid is the normal solvent used in the spinning processes. In wet spinning a strong solution of the polymer, which also contains inorganic salts, is spun through a spinneret into weak acid or water. In this bath the salts leach out. In the dry spinning process the salts are more difficult to remove and this process is only used to produce the weaker metaaramid fibres. In both processes post treatment of the fibres by additional drawing is used to optimise fibre properties. Aramid products are available as filament yarn, staple fibre or pulp. Properties and End-Uses Some of the main end-uses for meta-aramids are protective clothing, hot gas filtration and electrical insulation. Para-aramids are used to replace asbestos in brake and clutch linings, as tyre reinforcement, and in composites such as materials for aircraft, boats, high-performance cars and sports equipment. Members of police forces and armed forces wear anti-ballistic aramid apparel. Elastane yarns are characterised by their ability to recover from stretch. BISFA describes them as "a fibre composed of at least 85% by mass of a segmented polyurethane which, if stretched to three times its unstretched length, rapidly reverts substantially to the unstretched length when the tension is removed". Although elastane was first synthesised in 1937, it was not commercialised as a fibre until 1958. Properties and End-Uses In addition to their remarkable stretch and recovery properties, elastanes resist perspiration and cosmetic oils, are easily washable, are dyeable and have moderate abrasion resistance. Elastane yarns are often covered with another fibre before use. This provides more bulk and improves abrasion resistance. The main end-uses for the yarns are garments and other products, where comfort and/or fit are important. 21 22 There are many inorganic fibres, including glass, carbon, metal and ceramic. They are used particularly in the industrial fibre sector. Two important inorganic fibres are described in this section. Glass is the most important inorganic fibre. Production It is produced by melting glass pellets in an electric furnace at around 1500 degrees C. The molten glass passes through small holes in a plate at the base of the furnace. After cooling in air it is wound up on a package. Alternatively it can be spun centrifugally to form a web. Properties and End-Uses There are several types of glass fibre produced. They have in common high moduli, high rot resistance, low moisture uptake, are brittle and have low breaking extensions. Glass is used extensively for insulation in the form of a felt and also for reinforcing plastics to make boats, caravans, automobile parts etc. Other lesser uses are flame-resistant curtains and décor fabrics. The BISFA definition of a carbon fibre is "fibre containing at least 90% by mass of carbon obtained by thermal carbonisation of organic fibre precursors". Production The common precursors used to make carbon fibres are polyacrylonitrile(PAN) and pitch. If pitch is used the process consists of extrusion, oxidation and graphitisation. If PAN is used, a tow is oxidised, then carbonised followed by graphitisation. Properties and End-Uses Carbon fibres are characterised by having high moduli and high strength, especially when embedded in a matrix such as epoxy resin. They are also brittle and have a low density. The main end-uses are as reinforcement fibres in composites for the aircraft and aerospace industries and sports goods. 22 23 Figure 3. A classification of fiber based on diameter (cited from New Millennium Fibers, 2005 Woodhead Publishing Ltd.) 1.2.1 Industrial fiber The classification of fiber on the basis of its use is available and the industrical fibers are usually the fibers currently broadly applied in different industrical areas, e.g. for , 1.2.2 Textile fiber The textile fiber means the application 1.2.3 Military fiber 1.2.4 Synthetic fiber 23 24 1.2.5 Natural fiber Fibers classified as natural fibers are vegetable, animal, or mineral in origin. As the name implied, the vegetable fibers are usually derived from plants and the principal chemical components are cellulose, which has been also referred to the cellulosic fibers and which are basically bound to another natural phenolic polymer, lignin. With respect to these two main natural polymers, the vegetable fibers therefore are also referred to as the lignocellulosic fibers, except the cotton, which contain few lignin. The vegetable fibers are classified according to their source in plants as the bast or stem fibers, which are often used as the soft fibers for textile use; the leaf fibers, which are referred to as the hard fibers; and the seed-hair fibers, which is the most important vegetable fiber. 1.2.6 Organic fiber 1.2.7 Inorganic fiber 1.2.8 Smart fiber 1.2.9 High performance fiber High performance fibers are generally characterized by they presented remarkably high unit tensile strength and modulus as well as resistance to heat, flame, and chemical agents. The high performance fibers are usually contributed by rigid-rod polymers due to their liquid crystalline state, e.g. classified as lyotropic, such as the aramid Kevlar (DuPont), or thermotropic liquid crystalline polymers, such as Vectran (Celanese). The applications of these fibers are usually in the aerospace, biomedical, civil engineering, construction, protective apparel, geotextiles, and electronic areas. Poly(1,4-benzamide) (PBA) might be the first reported nonpeptide synthetic polymer to form a liquid crystalline solution. To obtain a liquid-crystalline solutions of poly(1,4-benzamide), the first step is to prepare the polymer in a proper solvent, e.g. N,N-dialkylamide, N,N-dimethylacetamide, and N,N,N,N-tetramethylurea. To obtain the high molecular weight, the lithium base, e.g. lithium hydride, lithium carbonate, or lithium hydroxide, is usually adopted in the polymerization solution during the reaction time posted 1–2 h. Thereafter, thermotropic polyesters, e.g. the acidolysis caused poly(ethylene terephthalate) by p-acetoxybenzoic acid (9), copolymer compositions that contained 40–70 mol% of the oxybenzoyl unit formed anisotropic, turbid melts which were easily oriented. Polyesters such as poly(p-phenylene terephthalate), 24 25 which can form liquid crystalline phases, decompose at temperatures below the melting point. The first reported high performance fiber is the polyaramid fiber, which was made based on poly(m-phenylene isophthalamide). Though this fiber was not liquid crystalline, the commercialized name by DuPont as Nomex nylon in 1963 and changed to Nomex aramid in 1972. In the 1970s, Kevlar fiber was developed by DuPont by processing of extended chain all para-aromatic polyamides from liquid crystalline solutions produced ultrahigh strength, ultrahigh modulus. The greatly increased order and the long relaxation times in the liquid crystalline state compared to conventional systems led this fiber has highly oriented domains of polymer molecules. 1.2.10 Special use fiber 1.2.11 Super fiber Generally, the super fibers are those fibers with remarkable mechanical properties, e.g. the strength more than ca. 2 GPa, and the elastic constant more than ca. 50 GPa. Notable, these units are absolutely different than the traditional fibers due to the latter represented in units of cN/dtex (centi-Newton/deci-tex). The value in cN/dtex represents the load per unit line density. On the other hand, the value in GPa represents the load per unit sectional area and is larger than that in cN/dtex. For example, the Para-aramid superfiber has a strength of 20 cN/dtex and elastic constant 500 cN/dtex (density = ca. 1.44 g/cm3) corresponds to 2.9 GPa and 72 GPa, respectively. 1.2.12 Dietary fiber 1.2.13 Medical fiber 25 26 1.2.14 Bio-fiber 1.2.15 Nano-fiber 1.2.16 Optical fiber Optical fiber transmits light where the refractive index varies in the radial direction. Fiber itself is one-dimensional, but light cannot be transmitted unless the structure is two-dimensionally controlled. Optical fiber is a powerful tool to transfer a large amount of information quickly, and plays a key role in supporting today’s information technology society. A fine optical fiber like a hair can transmit information equivalent to 6000 telephone circuits. Although the cost of optical fiber is higher than copper wire, the optical fiber is lighter in weight, higher in capacity and lower in the transmittance loss. An optical fiber is a fine filament, 0.1 mm in diameter, and transmits 95% of input light as far as 1 km. An optical fiber has a two-layer structure of core and clad. A core part is composed of the material with a high refractive index, and a clad part with a low refractive index. Light input in the core part 1.4 Importance of fiber science and technology 26 27 Figure 2-3. A summary of the use of fibers in today (cited from New Millennium Fibers, 2005 Woodhead Publishing Ltd.) Recommending reading [1] M. Harris, ed., Handbook of Textile Fibers, Harris Research Laboratories, Inc., Washington, D.C., 1954. [2] J. G. Cook, Handbook of Textile Fibers, Vol. I: Natural Fibers, 5th ed., Merrow Publishing, Durham, N.C., 1984. [3] Franz Fourne, Synthetic Fibers, Machines and Equipment, Manufactory, Properties. Hanser Publisher, Munich, Germany, translated by Dr. Helmut H.A. Hergeth, Raleigh, NC, USA, and Ron Mears, Obernburg, Germany. 1998. [4] J W S Hearle, High-Performance Fibres.CRC Press, Woodhead Publishing Ltd. Abington Hall, Abington, Cambridge CB1 6AH, England.2001. [5] Tatsuya Hongu, Glyn O. Phillips and Machiko Takigami. New millennium fibers. CRC press. Woodhead Publishing Limited. 2005. [6] J. Vincent Edwards,Gisela Buschle-Diller,Steven C. Goheen. Modified Fibers with Medical and Specialty Applications. Springer, 2006. [7] Roland Beyreuther, Harald Brünig, Dynamics of Fibre Formation and Processing, Modelling and Application. Fibre and Textile Industry. Springer, 2007. 27 28 2. Methods for fiber spinning 2.1 Physical methods 2.1.1 Melt spinning Figure 2-4. A scheme of the melt spinning process (cited from ) 2.1.2 Wet spinning In wet spinning, the polymer solution was prepared in a suitable solvent then be extruded as a fiber into a coagulation bath containing a nonsolvent. The limits of polymer concentration in the spinning solvent are determined by polymer solubility and solution spinning pressure limitations. The polymer concentration used for wet spinning is lower than that in dry spinning due to the solution spun at lower temperatures. Of the wet spinning, the spinneret was submerged in the liquid coagulation bath, and the emerging filaments are coagulated in a precipitating bath or a series of baths of increasing precipitant concentration. During the short residence time in the coagulation bath, the fiber formed and its structure taken shape as a result of complex. In this process, the counter-diffusion of solvent and nonsolvent and phase separation of the polymer would be taken place. 28 29 Figure 2-5. A scheme of the wet spinning. 2.1.3 Solution dry spinning During the solution dry spinning, the polymer spinning solution is initially metered at a constant temperature by a precision gear pump through a spinnerette into a cylindrical spinning cell 3–8 m in length. Heated cell gas, made up of solvent vapor and an inert gas, e.g. nitrogen, is therefore introduced at the top of the cell and passed through a distribution plate behind the spinnerette pack. Since the cell gas and cell walls are maintained with high temperature, this leads the solvent evaporation rapidly from the filaments in the spinning cell. The spinning solvent is followed condensed from the cell gases, purified by distillation, and returned for next run. A scheme of the solution dry spinning process was shown in below. 29 30 Figure 2-6. A scheme of the solution dry spinning process.(cited from Encyclopedia of Polymer Science and Technology. Vol. 6, p267, John Wiley & Sons, Inc. New York, 1993,) 2.1.4 Solution wet spinning In solution wet spinning process, the spinning solution is initially pumped by a precision gear pump through spinnerettes into a solvent–water coagulation bath. Then, the same process was done as the same as in the dry spinning to form the filament. At the exit of the coagulation bath, filaments are collected in bundles of the desired tex, and a false twist was usually employed at the bath exit to give the multifilament bundles a more rounded cross section. After the coagulation 30 31 bath, the multifilament bundles are counter-currently washed in successive extraction baths to remove residual solvent, then dried and heat-relaxed, generally on heated cans. For this process, the spinning solvent is generally recovered by a two-stage process in which the excess water is initially removed by distillation followed by transfer of crude solvent to a second column where it is distilled and transferred for reuse in coming fiber manufacture. A description of the spinning process was schemed in below. Figure 2-7. A scheme of the solution wet spinning process (cited from Encyclopedia of Polymer Science and Technology. Vol. 6, p267, John Wiley & Sons, Inc. New York, 1993) 2.1.5 Gel spinning 2.1.6 Liquid crystal spinning 2.1.7 Electrospinning 2.1.7.1 General electrospinning Electrospinning is an interesting technique for spinning polymers. Generally, this spinning method can offer fiber with excellent opportunity for designing its surface morphology and porosity to provide the most appropriate interface for biomedical applications. This process can be also regarded as a variation of the electrospraying. During the electrospinning, the solution viscosity, surface tension, conductivity, applied voltage and current are of importance. Polymer from a solution or melt can be deposited as fibrous material by charging the liquid, applying 5-30 kV. and ejecting it through a nozzle onto an oppositely charged grounded target as Figure ? described. Basically, an electrospinning system consists of a high-voltage DC supply, a grounded electrode, a nozzle system with diameter controls, and a fixed or rotated target to which the spun fiber could be adhered. The electrospinning is a simple way for producing nano-size filament by properly controlling 31 32 the polymer concentration and/or surface tension of the solution. The nozzle diameter can be in the range of 0.05 μm to a few microns. In general, the fibers was spun onto nonwoven structures which are porous and have high surface area. With respect to this property, the electrospun nonwoven structure is thus availably to provide scaffolding for tissue engineering. Figure 2-8. A scheme of the electrospinning. 2.1.7.2 Co-electrospinning Figure 2-9. Schematic illustration of the core solvent evaporation process from the co-electrospun fiber. (small 2007, 3, No. 6, 1064 – 1073) 2.1.7.3 Gas-jet/electrospinning 32 33 Figure 2-10. Schematic of the gas-jet/electrospinning equipment. Figure 2-11. Schematic of the spinneret of the gas-jet/electrospinning equipment. (J Appl Polym Sci 107: 909–917, 2008) 33 34 Figure 2-12. SEM images and diameter distributions of PES fibers gas-jet/electrospun from (a) 10, (b) 18, (c) 20, (d) 22, (e) 25, and (f) 27 wt % PES solutions. The original magnifications were (a) 5003 and (b–f) 50003. 34 35 2.1.8 Bi-components spinning 2.1.9 Reaction spinning Akron Milicron 1-1/2 inch extruder Banbury external mixer 35 36 2.1.10 Centrifugation spinning 2.1.11 Interfacial polycondensation spinning 2.1.12 Laser associated spinning 2.1.13 Coaxial spinning 2.1.14 Dry spinning By heating solution of polymer in solvent then extruded into a hot stream of gas, which can be also continuously heated during its passage, and evaporating the solvent, the fiber therefore reacts to the loss of solvent by passing through a gel to a solid state. In such process, the fiber surface hardens initially while the solvent still diffusing out from the interior, and a cylinder under external over-pressure is formed. The internal pressure decreased with the distance increased from the spinneret and the surface suddenly collapsed to rise to a round, oval or serrated, deformed cross-section, depending on the polymer. Of the dry spinning process, the exact spinning conditions, e.g. concentration, viscosity, temperature, spinning gas flow rate etc., are polymer-and solvent specific. On the use of the dry-spinning technique, the recovery of the solvent is absolutely essential, while the recovery of the spinning gas might be also advisably. 36 37 Figure 2-10. A scheme of the dry spinning. 2.1.15 Dry jet-wet spinning In wet spinning, the as-spun fiber always has voids that cause deterioration of the weak mechanical properties for fiber, probably the fibrillation and low transparency occurred easily. Since the formation of the voids can be controlled in a minimum by extruding the dope stream from a dry jet, and subsequently followed by coagulation as in conventional wet spinning, such process is regarded as the dry jet-wet spinning. The dry-jet-wet spinning can allow the stress relaxation for polymer chains in the air gap of the orientation produced in the spinneret to cause the spun fiber less oriented and more uniform than that from the immersed jet. This permits orientation by subsequent drawing and gives fibers with higher tenacity. This novel fiber spinning technique was schemed in Fig.? . Figure 2-11. A scheme of dry-jet-wet spinning. 2.2 Chemical methods 2.2.1 Emulsion spinning 2.2.2 Polymerization method In a typical fabrication, a cationic surfactant decyltrimethylammonium bromide (DeTAB) or dodecyltrimethylammonium bromide (DoTAB) was dissolved in distilled water to form micelles as the nanoreactors. Table 1 summarizes the chemical structure, critical micelle concentration (CMC), and micelle aggregation number (n) of the surfactants, DeTAB and DoTAB. The micelle aggregation number is the number of 37 38 surfactant molecules that form a micelle. The CMC of cationic surfactants decreases with increasing hydrocarbon chain length, owing to the enhanced hydrophobic interactions of the chains. Spherical micelles form between the CMC and the concentration for sphere-to-cylinder transformation (0.5 and 0.35 M for DeTAB and DoTAB, respectively; Fig. 1a).[8] Under these conditions, the strength of the hydrophobic interactions exceeds that of the electrostatic repulsions of the ionic head groups, which stabilizes the formation of micelles. Hydrophobic alkyl chains remain tangled in the micelle. Acrylonitrile (AN) monomer was then added dropwise to the micelle solution. The hydrophobic monomer could penetrate into micelles, hence the micelles swelled significantly. AN monomers have poor solubility in water, whereas they are soluble in organic solvents such as N-methylpyrrolidone (NMP).[9a] Therefore, the AN monomers were likely to be introduced into the hydrophobic inner spaces of the micelles. Theoretically, it is also very difficult for the AN monomer to stay free in the microemulsion system, owing to the high surfactant ratio (i.e., the large number of micelles). Polymerization of AN without surfactant produced sub-micrometer-sized (500 nm) PAN particles with a broad size distribution.[9b] Subsequently, the monomers inside the micelles were polymerized using the redox initiators cerium sulfate and nitrilotriacetic acid (NTA; Fig. 1b). Polymerization of AN readily occurred, owing to the relatively high oxidation potential of cerium sulfate. After 30 min polymerization, an amount of FeCl3 was introduced, and the additional polymerization proceeded with magnetic stirring (Fig. 1c). The introduction of salt promoted micelle growth from spherical (several nanometers) to bigger rodlike or cylindrical micelles, because the addition of salt lowers the charge density around the micelle surface and hence will decrease the entropic penalty in transforming the micelles.[10a] Figure 2a and b shows the light-scattering correlation functions and size distributions, respectively, of spherical (Fig. 1b) and rodlike (Fig. 1c) micelle solutions. In the correlation function of the spheres, one inflection point is clearly observed. This indicates that the spherical micelles exhibited only translational motion. Two inflection points existed in the case of rodlike micelles, due to the change in molecular motions of micelles—possibly rotational motion.[10b,c] While the spheres showed one peak attributed to the diameter of the micelle, the rodlike micelles exhibited two peaks associated with the radius and length of the micelle.[10b] In addition, the viscosity of the rodlike micelle solution (Fig. 1c, 1.6 cps; 1 cps = 1 mPa s) increased compared with that of the spherical micelle solution (Fig. 1b; 1.4 cps).[10b,d] These facts qualitatively support the fact that the sphere-to-rod micelle conversion was successful owing to the addition of salt. In our experiments, the diffusional degradation/coagulation of the micelles was negligible, for the following two reasons. First, the size of the PAN-embedded micelles was not significantly affected by the micelle coagulation, because PAN cannot grow infinitely by the penetration of monomer into micelles.[10e] Second, the diffused monomer cannot easily swell the prepared PAN nanoparticles in the micelles. Judging from these facts, it is clear that the intermicellar exchange of AN monomer did not modify the micelle structure significantly. When the salt was added prior to polymerization, PAN nanoparticles with a broad size distribution were obtained. Similarly, salt addition after 5 h polymerization produced PAN nanoparticles with an even broader size distribution. The prepared polymer nanofibers were retrieved by precipitation in an excess of methanol to remove surfactants and initiators. Figure 3a presents the transmission electron microscopy (TEM) image of PAN nanoparticles produced after polymerization for 30 min. The average diameter of the PAN nanoparticles was approximately 20 nm and showed a nar- row size distribution. It was very difficult to fabricate PAN nanoparticles, because PAN is not soluble in the monomer, owing to strong molecular interactions.[10e] However, it was possible to prepare PAN nanoparticles with a narrow size distribution owing to the high oxidation potential of the initiators. The preparation of PAN nanoparticles with a uniform size was very important, because the individual PAN nanoparticles act as a lattice crystal (building block) for nanofiber formation. This is therefore also important for the production of nanofibers with a narrow size distribution. The concentration required for transformation of spheres to cylinders drops significantly, possibly by an order of magnitude, on salt addition.[10e,11] In this way, 38 39 spherical micelles containing PAN nanoparticles were able to transform to cylindrical micelles after the addition of iron(III) chloride. Consequently, the self-assembled PAN nanoparticles generated a nodular nanofiber structure (Fig. 1c). The additional polymerization of the residual monomer inside the cylindrical micelles then produces the smooth surface of the PAN nanofibers. The average diameter of the PAN nanofibers is about 25 nm, which is slightly higher than the average diameter of PAN nanoparticles, and they are reasonably monodisperse (Fig. 3b). This diameter increment indicated the successful additional polymerization outside PAN nanoparticles after the addition of salt. Energy dispersive X-ray (EDX) spectroscopy showed that the PAN nanofibers consisted of carbon (72.5 %), nitrogen (23.5 %), iron (3.6 %), and traces of initiator (sulfur and cerium). The atomic ratio of carbon/nitrogen was 3.34, which coincides with the theoretical atomic composition of PAN. The successful fabrication of PAN nanoparticles and nanofibers was further confirmed by FTIR and UV-vis spectroscopies. The FTIR spectra of PAN nanoparticles and nanofibers (Fig. 4a) showed a PAN C_N stretching band at 2244 and 2236 cm–1, respectively. The sharp peak at 1620 cm–1 from the C_C stretching mode in the AN monomers completely disappeared after polymerization. The FTIR analysis demonstrates the successful polymerization of the AN monomer. The UV-vis spectra displayed a single absorption band at 290 nm, which is the characteristic band of PAN (Fig. 4b). It is remarkable that the vibrational band for the C_N stretch in PAN nanoparticles, observed at 2244 cm–1, down-shifted to 2236 cm–1 in the case of PAN nanofibers. Simultaneously, the EDX analysis revealed the presence of embedded iron. Therefore, it was inferred that the vibrational motion of the CN group was restricted by coordination with embedded iron, which supports the presence of coordination between iron and the cyano groups in PAN. Figure 11. Overall synthetic procedure of polymer nanofibers using microemulsion polymerization.( Adv. Funct. Mater. 2006, 16, 1400–1406) 39 40 Figure 3. TEM images of a) PAN nanoparticles with an average diameter of 20 nm and b) PAN nanofibers with an average diameter of 25 nm obtained with 0.3 M DoTAB.( Adv. Funct. Mater. 2006, 16, 1400–1406) Figure 4. a) FTIR and b) UV-vis spectra of PAN nanoparticles and nanofibers obtained with 0.3 M DoTAB. 40 41 Figure 10. a) SEM image of carbonized PAN nanofibers. b) Raman spectra of carbon nanofibers as a function of carbonization temperature. Recommending reading [1] [2] [3] 2 Fiber characterization methods 3.1 Morphology The morphology of textile fibres can be complex, involving crystallinity, molecular orientation and the grosser morphology determined by the physical conditions and mode of phase separation as the fibre is spun, in Case Study 2.1, from solution into a non-solvent. In order to understand the reliance of the final product on the spinning conditions, it is necessary to first be aware of the nature and possible routes to phase separation. The resolving power (the smallest distinguishable separation of two point objects) of any microscope is related to the radiation wavelength by: 41 42 where the numerical aperture NA is given by Figure 3-1. Resolving power where n is the refractive index of the medium through which the beam travels before reaching the specimen. Another important parameter is the depth of focus, T, which is related to the beam aperture, , and the resolving power, : Figure 3-2. Depth of focus 3.1.1 Optical microscopy The useful magnification, M, of an optical microscope is related to the resolving power of the eye - typically of the order of 0.1mm: Orientation and Birefringence Amorphous polymers possess an isotropic refractive index and when placed between two crossed polarisers in a light microscope no light is transmitted. An oriented polymer exhibits 42 43 anisotropic refractive indices as, molecularly, the refractive index along the chain is generally different to that perpendicular to it. The effect of the orientation is thus to rotate the plane of polarised light and an image is observed when the oriented specimen is placed between cross polarisers. The effect is termed birefringence and is exhibited also by crystalline polymers. Orientation birefringence disappears at and crystalline birefringence disappears at . In uniaxial orientation the birefringence, , is simply the difference in refractive index, n, in directions parallel and perpendicular to the orientation direction. The actual measurable characteristic of the material is < > where is the angle between the statistical segment and the direction of the force inducing orientation; the brackets denote an average. The Hermann's orientation function, where , is defined as: = 0 for unoriented material and 1 for perfectly oriented and n can be shown to be: where is the average refractive index is the number of network chains per unit volume and are the polarisabilities parallel and perpendicular to the statistical segment axis; if these characteristics of the material are known then can be determined from the birefringence. The orientation present in a quenched, drawn material can be determined by the compensator method. This involves placing a wedge of anisotropic material into the light path before the analyser. Altering the thickness of the compensator introduces different degrees of retardation into the beam and when the specimen retardation is compensated the field becomes dark. The optical retardation, R, imposed on a light beam by the specimen is defined as: where d is the sample thickness is the monochromatic incident light wavelength: 43 44 Figure 3-3. Measuring birefringence The polarisation directions of the polariser and analyser are + and - 45 to the draw direction. 3.1.2 Electron microscopy Electron microscopy is analagous to light microscopy where the light beam is replaced by a beam of electrons. The electron beam is focussed by a series of electrostatic or electromagnetic lenses. Because electrons are scattered by air the beam path must operate in a vacuum - for this reason volatile specimens are not suitable for electron microscopy. The contrast is provided by differences in electron density within the specimen, often these may not be sufficient to give a great enough difference in scattering power to allow different material domains to be distinguished and etching or staining techniques must be employed. The resolving power of the electron microscope, is given by: The de Broglie wavelength of the electron beam is where h is Planck's constant and mv is the electron momentum. The velocity, v, of the electron can be determined from the accelerating voltage: where e is the electron charge U is the accelerating voltage m is the electron mass and is then given by where U is in volts. A relativistic velocity correction yields somewhat higher values of . Lens aberrations lead to point resolving powers of tenths of nanometers. If the resolving power of the microscope is 0.2nm then the useful magnification, M, is given by (for TEM): 44 45 For n=1 and small , the depth of focus is given by: When these high energy electrons interact with the specimen, X-rays may also be emitted and can themselves be used as an analytic tool. Electron diffraction patterns can be obtained where the shorter wavelength results in reflections at smaller angles. The strong interaction of the electron with an atom yields very intense patterns thus reducing the exposure time compared to a conventional X-ray pattern 3.1.3 Scanning electron microscopy In scanning electron microscopy the beam sweeps the surface of the sample synchronised with a beam from a cathode ray tube. Backscattered electrons from below the surface of the specimen modulate the intensity of the beam from the cathode ray tube. Secondary electrons may arise from a thin surface layer.The useful magnification of SEM is limited by the size of the scanned surface area in comparison to the size of the screen on which the image is projected. Back scattering requires high accelerating voltages and heavier elements and is therefore more suitable for metals than for polymers. Resolution depends on the size of the electron beam and on the scattering volume within the sample but the depth of focus is large as no lenses are employed in image formation. Preparation of samples may involve surface deposition of a thin conductive layer to prevent charging of the surface. This conductive layer masks surface features to some extent, limiting resolution to about 10nm. Internal structure can be investigated by cleaving the sample, often at reduced temperature. 3.1.4 Transmission electron microscopy The elements which form a TEM are analagous to those in a light microscope. Electrons are fired from an electron gun, passing through a condenser lens and through the specimen; the emergent beam is refocussed by an objective lens and a projector lens forms an image. Sample preparation techniques are many and various and all involve the production of a specimen which, in the case of polymers, should be less then 0.1mm thick to give an acceptable signal to noise ratio. Replicas can be made by applying a thin film from solution or by vapour deposition; surface morphology can be investigated at a higher resolution than SEM. Internal features can be investigated either by replication of fracture surfaces or by ultramicrotoming. Contrast can be enhanced by selective staining e.g. osmium tetroxide is used with polymers containing C=C double bonds. 45 46 3.1.5 Scanning transmission electron microscopy Analagous to SEM in construction electrons scattered in transmission are used to modulate a CRT beam and thus form an image. The advantages stem from the elimination of lenses in image formation and the reduction of electron beam damage to an object which is scanned by the electron beam. 3.1.6 Atomic force or scanning probe microscopy The development of the Atomic Force Microscope (AFM) was announced in 1986 (Reference 36). The instrument allows imaging of surfaces on a nanometre scale where specimens do not need to be electrically conductive as is the case for the scanning tunneling microscope. The AFM measures the attractive or repulsive forces between a probe tip and the sample surface. The probe tip is attached to a flexible cantilever and is made to scan the surface of the sample. Originally two modes were in use - contact and non-contact modes: 1. In contact AFM the probe tip is constantly in contact with the sample surface. Surface contours bend the cantilever. The cantilever is adjusted to maintain a pre-determined bending and the adjustment required is monitored as a function of position on the surface. Disadvantages of this method include the attractive effect of adsorbed liquid on the sample surface and electrostatic forces which introduce both compressive and shearing forces as the tip is dragged over the sample surface. These problems can make measurement difficult and can damage the surface of soft samples. 2. In non-contact mode the tip does not contact the surface but senses the weak Van der Waals force between the tip and the sample. The cantilever is usually vibrated at small amplitude and experiences a shift in its resonant frequency due to the interactive forces. The major disadvantage of this technique is the nature of the Van der Waal's force which is very weak and highly localised. The Tapping Mode technique is closely related to the contact mode but seeks to minimise its disadvantages. In particular the shear force is eliminated by tapping the sample surface with the probe as it scans so that it is not dragged across the surface. A further refinement is the Near Field Scanning Optical Microscope (NSOM) which funnels visible light through the tip onto the sample surface. The light is either transmitted through the sample or reflected from the surface. The probe must be kept at a constant distance from the surface for a true image to be obtained. 3.2 Thermal properties 3.2.1 DSC - Differential scanning calorimetry DSC can identify the following: 46 47 transition temperatures such as and crystallisation temperature heat capacities heats of transition specific heats decomposition temperature melt enthalpies compatibility of blends degree of crystallinity from the heat of fusion A reference sample and the experimental sample are heated separately but in parallel according to a linear temperature programme. The two heaters maintain the two samples at identical temperatures. The power supplied to the two heaters to achieve this is monitored and the difference between them plotted as a function of reference temperature which translates as a recording of the specific heat as a function of temperature. As the reference temperature is increased or decreased and the experimental sample approaches a transition the amount of heat required to maintain the temperature will be greater or lesser depending on whether the transition is endothermic or exothermic. The heating rate has important effects on the results, for example a fast heating rate will delay the onset of . 3.2.1 DDSC-Dynamic Differential Scanning Calorimetry A relatively new technique which applies a sinusoidal thermal program to the sample. The technique can separate processes which may occur simultaneously such as glass transition and crystallisation. Software and hardware upgrades are required for standard DSC equipment in order to be able to perform DDSC. 3.3 Mechanical properties 3.3.1 Tensile strength 3.3.1 Tensile testing -stress-strain experiments A typical tensile experiment designed to record a stress-strain curve requires the following elements: -a method of gripping a standard specimen - usually a dumb-bell shape - securely -a means of stretching the specimen at constant rate -a range of strain rates -a means of measuring strain, strain rate and load an ability to impose appropriate loads for the material under investigation e.g. an INSTRON tensile tester offers a choice of several different load cells and one would be chosen for a particular experiment dependent on the maximum load required - often the load to break; choosing 47 48 a load cell with too low an upper limit could result in damage to the load cell; choosing one with two high an upper limit reduces the resolution of the data set. A Young's' modulus can be determined (for a given strain rate) for a polymer by determining the slope of a stress-strain curve at low (<1%) strain. Stress relaxation data are recorded by rapidly imposing a strain on the specimen and monitoring the load requird to maintain the strain. Stress-strain experiments can also give a measure of the cross-link density in a rubber. At constant temmperature the stress (force/unit unstretched cross-sectional area) is proportional to where is the extension ratio and is the molecular weight between entanglements. The specimen is clamped in the Instron and stabilised at the test temperature. Application of a force to stretch the specimen and monitoring of the resultant yields a value for . Creep experiments can be performed in specially designed stand-alone chambers which can vary thermal and atmospheric conditions such as humidity and nature of the surrounding gas. The experiment simply involves attaching a load to the specimen and monitoring the extension as a function of time and other experimental conditions. The tensile properties of the fibers in terms of the breaking tenacity, percentage of breaking elongation and Young’s modulus were determined using an Instron (Model 4000, Instron, Norwood, MA) tensile testing machine. A gauge length of 25 mm and a crosshead speed of 18 mm/min were used for the testing. Approximately 100 fibers were tested for their tensile properties. The moisture regain of the fibers was determined as the ratio of the difference in the dry weight of the fiber to the weight of the fibers conditioned for 24 h at 21℃ and 65% relative humidity. Flexsys, Tensometer 2000 48 49 3.3.2 Dynamic mechanical experiments Polymers can show all stages of behaviour from viscous to elastic depending on temperature and frequency. At high temperatures and low frequencies they are more likely to show viscous behaviour while elastic behaviour dominates at low temperatures and high frequencies. Rapid changes in properties can occur if resonances or relaxations are stimulated at particular frequencies and temperatures; these include the glass transiton. Dynamic mechanical experiments subject the specimen to a sinusoidal strain and monitor the resulting sinusoidal stress characterised by the storage modulus, loss modulus and the phase difference between the two - a measure of the damping of the polymer. Several methods are available covering a wide range of frequency and temperature (Reference 7): free vibration method - the torsion pendulum: specimen is clamped vertically and the lower clamp is twisted and released; the resulting damped oscillations of the specimen are monitored. Vibration frequency depends on the stiffness of the material which is temperature dependent. forced vibration - for investigating frequency and temperature effects: a sinusoidal strain is 49 50 imposed, low enough to comply with linear viscoelastic theory 3.3.3 Ultrasonic modulus measurements The ultrasonic immersion technique is a method for measuring the elastic properties of a material. The sample to be measured is placed between an ultrasonic transmitter and receiver (2.25MHz) in a temperature controlled water tank, and the time for an ultrasonic pulse to traverse the media between the transducers is measured. Both tensile and shear waves are propagated when the sample is rotated, and the resulting data relating the sound velocity in the sample to the angle of incidence allows the elastic properties of the sample to be determined. When perpendicular to the direction of the ultrasonic pulse only a tensile wave is generated in the sample. When rotated, refraction at the front surface can produce a shear wave. The velocities of both types of wave are measured over a range of incidence angles. For more details of the technique see the original work of Read and Dean (Reference 42), the thesis of Lord (Reference 43), and the papers by Dyer et al (Reference 44) and Woolf (Reference 45). The technique determines the stiffness constants of a material, to measured stresses , which relate applied strains by the reduced Hooke's Law: where i= 1,2,3 are tensile stresses and strains j= 4,5,6 are shear stresses and strains. This results in a series of six equations, conveniently expressed in matrix form, which, in the most general case, lead to 36 stiffness constants. In most materials there are varying degrees of symmetry which reduce the number of unknowns. For example, for most materials the symmetry axes of the sample correspond to the major property directions such that the application of tensile strains (j = 1,2,3) do not produce shear stresses and the application of shear strains do not produce tensile stresses. This results in the majority of the off-axis elements of the matrix being zero. The most complicated case likely to be encountered is for orthrhombic symmetry but with orthotropic properties: i.e. different properties along the three main structural axes. One example of this kind of material is an injection moulded glass filled composite. In this case the matrix of stiffness constants reduces to 9 independent values and is given by: A further simplification is for the material to have an isotropic plane in which the properties are the same in the two directions. A typical example would be a unidirectional fibre reinforced composite, which has an isotropic plane perpendicular to the main fibre direction. This form of 50 51 symmetry is termed transverse isotropy or fibre symmetry and has only five independent stiffness constants: The final case is for an isotropic material which has the same properties in all directions resulting in only two independent constants. A similar set of matrices exist which relate applied stresses to measured strains through the compliance constants which can be obtained by inverting the matrix: The compliance constants can be related to the more often quoted engineering constants, Young's Modulus E, shear modulus G, and Poisson's ratio . The aim of the ultrasonic immersion method is, therefore, to determine all of the independent stiffness/compliance/engineering constants for the particular material under test. 3.4 Rheological properties 3.4.1 Capillary rheometry Several instruments have been developed to measure rheological properties in shear. Each instrument has its own individual strengths and weaknesses. The capillary rheometer is capable of measuring rheological properties at shear rates which are comparable to many polymer processes. Figure 2-11 is a schematic view of a capillary rheometer. Figure 3-. Schematic view of a capillary rheometer (ref.79). Solid polymer is loaded into the reservoir at the top of the rheometer where it is melted at the desired temperature. After this melt time, the plunger is driven down forcing molten polymer through the capillary region. Measurements of the flow rate of material and the force required to move the plunger, corrected for frictional effects, allows the calculation of the material’s viscosity. Making a balance of forces (neglecting gravity) on a fluid element within the capillary yields eq. 8. (1) 51 52 where rz(r) is the shear stress along a surface of constant r in the z direction is the pressure gradient driving flow Thus, for a capillary of radius R, the shear stress at the wall (r = R) of the capillary, w, can be defined and related to the shear stress as follows. (2) From eq. 9 it can be seen that the shear stress in the capillary is a linearly varying function of position, regardless of the material being tested. That is, to this point no constitutive equations have been used. Assuming that fluid only flows in the z direction and that the “no slip” boundary condition applies (uz=0 @ r=R), and considering that the shear rate is simply the first derivative of the fluid velocity with respect to radial position, the following relations may be derived. (3) Equation 10 provides a relationship between _, , and . Thus if two of the quantities can be measured, the third can be calculated. In a capillary rheometer and _are obtained at the wall of the capillary through measurements of force on the plunger and flow rate respectively. Equation 10 can be used to obtain the Rabinowitsch equation. where a new term is introduced, the apparent shear rate, , which can be defined as follows. The term in parentheses in eq. 11 is commonly referred to as the Rabinowitsch correction. For Newtonian fluids _w and are equivalent because there is no dependence of viscosity on shear rate. However, for non-Newtonian fluids, w and are not equivalent. A shear thinning material will have a steeper velocity gradient near the wall, where viscosities are determined in capillary rheometry, as compared to a Newtonian fluid. Thus, errors may result if the Rabinowitsch correction is not applied. For a power law fluid, the Rabinowitsch correction factor becomes, where B is the power law exponent from eq. 5. Additional corrections can be made to capillary rheometry data to further improve accuracy. Figure 2-12 shows an additional affect which may cause error in measurements. At the entrance of the capillary in the reservoir, flowing fluid is rapidly forced from a large cross section into a much smaller cross section. This entrance effect leads to an additional pressure drop 52 53 which can be corrected for by using the method of Bagley (ref.12). The method requires that measurements be taken using two (ideally three or more) capillaries of varying L/D ratio. Additional errors in measurement may arise from the following: the finite amount of kinetic energy added to the system to accelerate the fluid in the reservoir, an additional pressure drop from friction between the plunger and reservoir walls, and from the small pressure dependence of viscosity. Finally, due to the elastic nature of polymer melts, some energy can be stored in the material as it exits the capillary, resulting in a pressure greater than atmospheric in the material at the outlet. This effect manifests itself visibly as die swell, and is due to normal stresses within the material. Normal stress data can be obtained from a capillary rheometer by measuring the amount of die swell. One author, Cogswell, has shown that elongational viscosity data can be obtained with an analysis of entrance effects. The entrance effects can be related to extensional viscosity because the entrance flow to the capillary will have an elongational component, as shown in Fig. 2-12(ref. 13). Experimental Procedure The speed of sound in the water tank held at a constant temperature - usually 30 - is first measured by monitoring the transit time as the separation of the transducers is changed. The relative positions of the transmitter and receiver are then set so that the travel time of the pulse is 100 . The sample is then inserted between the two transducers. It is adjusted to be perpendicular to the ultrasonic pulse by setting the transmitter to act additionally as a receiver and maximising the reflected pulse. The experiment consists of rotating the sample away from this position and monitoring the transit time of the tensile pulse and that of the shear pulse which appears as the sample is rotated. The thickness of the material at the transmission point is measured together with the material density. The experiment can be repeated forthe other axis by rotating the specimen in its plane: 53 54 Figure 8.10 Measurement of orthogonal moduli If a third experiment is necessary then the sample must be cut into strips and reassembled to give the correct aspect. ? Appendix: Stresses and Strains ? Tensile Deformation A few definitions are presented in relation to a body subjected to a stretching force. Figure 9.1 Schematic of body subjected to a stretching force The tensile or engineering strain, e, is defined as: 54 55 where is the initial length of the specimen and l the extended length. The degree or per cent elongation is defined as: The extension or draw ratio, , is defined as: The extension ratio will be less than unity for the two directions orthogonal to the stretching direction. Poisson's ratio, , is the ratio of lateral contraction per unit width to the longitudinal extension per unit length, in a simple tensile deformation: Usually it takes a value between 0.2 and 0.4; a value of -1 indicates no change in proportions, and a value of +0.5 indicates no change in volume. The tensile stress, , is given by: where F is the applied force and the initial cross-sectional area. Stress is related to strain via Young's modulus E: ? Shear Deformation Similar expressions can be obtained for shear: Figure 9.2 Schematic of shear The shear or rigidity modulus G is defined in terms of the shear stress (= assumed to be small and equal to dl/l): 55 and the shear strain 56 ? Hydrostatic Pressure The bulk modulus K is defined in terms of a hydrostatic pressure (equivalent to negative tensile stresses applied simultaneously in all three directions), and the volume V: where the bulk strain is . ? Relationships for Isotropic Bodies It is simple to demonstrate that for isotropic materials the bulk modulus is and the shear modulus can be related to Young's modulus, E, and Poisson's ratio , : ? Generalised Stress-Strain Relationships The simple engineering approach to mechanical properties is adequate for many purposes, especially when the systems under consideration are isotropic. Unfortunately, fibres and films are frequently anisotropic and a more formal approach is required. ? Stresses Stress is defined as a force per unit area and so all definitions of stress must be related to an area over which a force acts. Consider a body which is being stretched; how do we define stress within the body? We do this by imagining a cut through the point of interest and asking what force, , acting on the cut surface, would be required to keep everything in position. We then resolve this force into its component normal to the cut surface, tensile stress is given by and the shear stress by , and in the plane of the surface, .The . Clearly, the results of this procedure depend upon the orientation of our surface or, in other words, our choice of axes, since we make the cuts perpendicular to our chosen axes. 56 57 Figure ? Schematic of force on a ``cut`` surface within a body Generalised expressions for the components of stress and strain are thus obtained by consideration of an infinitesimal cube of material within a body (Reference 46): Figure ? Components of stress The cube's edges are parallel to the x,y,z axes and it is deemed to be in a state of equilibrium. The forces , and act upon the yz, zx, and xy planes respectively. In the absence of torque: and therefore only six independent parameters are required to define the state of stress - the three normal stress components . , , , and the three shear stress components , , and ? Strains Associated normal and shear strain components are , , , and , , . The engineering components of strain are defined purely by consideration of the relative displacement of points within the bulk rather than absolute displacement. The co-ordinates of the initial 57 58 positions of the two points are , , , and , , : Figure 9.5 Displacement of two points The two points are displaced to , , , displacements , , in , and the deformed , state. The relative are the quantities of interest and are related to the normal and shear components of strain. The expression for strain is related to the deformation i.e. the relative displacement of two points initially at co-ordinates , , , and , , and is a measure of the distortion, as distinct from rigid body translation or rotation. The state of strain can also be expressed as the components of a strain tensor, , defined as: The tensor, , and engineering, e, components of tensile strain are identical : where , , are the fractional expansions and contractions perpendicular to the respective planes xx, yy, and zz. In shear: where , , , are the components of shear strain in the respective planes xy, yz, xz, and ? Compliance and Stiffness The most general linear relationship between stress and strain assumes that each of the tensor components of stress is linearly related to all of the tensor components of strain and visa versa. This gives the most generalised form of Hooke's Law. Use of tensor notation yields expressions for the compliance and stiffness constants. The mechanical properties of an anisotropic solid can therefore be represented by a generalised Hooke's law relating the second rank tensors of stress and strain via fourth rank tensors, respectively: and , which are compliance and stiffness constants 58 59 An abbreviated nomenclature is used (Reference 46) to relate the six independent components of stress, , to the six independent components of strain, , where p and q go from 1 to 6: For instance, in a specimen of fibre symmetry the compliance matrix reduces to: where z has been chosen as the axis of symmetry and the number of independent constants has been reduced to five. Young's modulus is then and Poisson's ratio: 3.4.2 Shear modulus 3.4.3 Shear strength 3.4.4 Density 3.4.5 Tenacity 3.4.6 Elongation 3.4 Light scattering The calculation of molecular weight by light scattering is well-established and relies on the variation of scattering with polymer concentration and scattering angle, where the polymer has a different refractive index to the solvent. The intensity of light scattered by a particle, angle to the incident beam of intensity where is the Rayleigh ratio, , at an is given by: is the distance between the detector and the scattering volume, is the polarisability of the particle and is the wavelength of the light. 59 is related to refractive 60 index and so changes in refractive index result in changes in the scattered light intensity. It was shown by Debye (Reference 33) that for polymer solutions: where K is a function of the refractive index, n; dn/dc is the refractive index increment or concentration dependence of the refractive index; the size and shape of the molecule, and polymer-solvent interactions and related to molecular weight is the particle scattering factor related to is the second virial coefficient, a measure of , the Flory interaction parameter. The weight average can be determined via a series of experiments over a range of concentrations where : where is the number of molecules of molecular weight the summation is over i, the complete range of possible molecular weights. Measurement of molecular weight by freezing point depression, boiling point elevation or osmotic pressure would yield the number average molecular weight defined as: Flow properties are affected by the molecular weight distribution or polydispersity - the ratio of weight average to number average molecular weight. The closer the ratio is to unity the more nearly monodisperse is the system.Another average is the viscosity average molecular weight which is determined by viscosity techniques (Reference 14). Single phase systems i.e. of uniform refractive index, do not scatter light. Two phase systems in which the two phases have different refractive indices do scatter light. Light scattering is, therefore, a useful technique for observing polymer-polymer compatibility. As a blend system proceeds from one phase through phase separation to a two phase system the system will begin to scatter light and to turn cloudy. Scattering patterns for binodal and spinodal separation are different and can be followed by light scattering. 3.5 X-Ray techniques X-rays are generated when a high energy beam of electrons strike a target. The incident electrons either radiate X-ray photons as they are rapidly decelerated (Bremsstrahlung -`braking radiation') or eject electrons from the innermost shells of the target atoms. In the former case the X-rays have a range of wavelengths; in the latter case electronic rearrangement within the target atoms results in the emission of X-rays of wavelengths characteristic of the target material. X-rays are scattered by variations in electron density.Where the electron clouds are ordered with respect to each other the scattered X-rays can interfere according to the Bragg condition to produce a diffraction pattern characteristic of the ordering. Bragg's Law states: 60 61 where is the X-ray wavelength d is the spacing between the atomic planes is the angle between the X-ray beam and the atomic plane Figure 8. X-ray reflection from atomic planes Constructive interference occurs for integer values of n. By measuring for a known wavelength the Bragg spacing d can be determined. X-ray diffraction can be used as a means of estimating the degree of crystallinity by separating the components of a diffraction pattern due to crystalline and amorphous regions.This estimate may well differ, however, from an estimate obtained by a different technique such as density measurements, due partly to a contribution to the amorphous halo by diffuse scattering from crystalline regions (Refererence 35). If a pure amorphous specimen of a crystallisable polymer can be prepared, and the crystalline reflections do not interefere with the amorphous halo, then the scattered intensity is assumed to be simply the sum of the scattered intensities from the crystalline and amorphous regions and the degree of crystallinity can be calculated from their relative magnitude. Wide-angle diffraction is able to access correspondingly smaller features such as chain packing in crystalline regions. On the smallest scale, the technique is sensitive to correlations between nearest neighbours in polymer chains and to correlations in the distances separating chains. It is therefore possible to investigate both tacticity and conformation as well as orientation in drawn polymer fibres. If, for example, an amorphous polymer is cold drawn, the typical duffuse halo can develop increased intensity in the diffuse halo perpendicular to the draw direction and reduced intensity elsewhere - Figure 8.3. This is due to the molecular orientation conferring a degree of order in the material. A semi-crystalline polymer will typically exhibit an amorphous halo and several sharp concentric rings associated with the randomly oriented crystalline regions. If such a polymer is uniaxially oriented the x-ray diffraction pattern - called a fibre pattern - will develop symmetrically spaced high intensity spots and arcs characteristic of regular features in the crystalline regions. 61 62 Figure 8. Effect of orientation on a) the amorphous halo and b) concentric ring pattern To obtain fibre patterns of a uniaxially oriented polymer, a number of fibres must be stacked to give a sufficient scattered intensity. Notice in Figure 8.3 that sharp arcs are not present on the meridian. In order to obtain information about spatial correlations in this direction the specimen must be tilted so that the deformation axis is not perpendicular to the plane containing the incident and scattered X-ray beam. Patterns are generated over a range of such inclination angles to give a complete picture of the orientation state of the polymer. More complex orientation states e.g. biaxial, require many experiments exploring diffraction as a function of variation in the azimuthal angle as well as the angle of inclination. This 3-d mapping is known as a pole figure. In small angle X-ray scattering (SAXS) fluctuations in electron density over dimensions of typically 3 - 40nm contribute to the diffraction patterns. Thus the long period of crystalline lamellae can be investigated along with information on the initial stages of crazing and microporosity in fibres. The electron density is different not only for crystalline and amorphous regions, but also in microvoids within the polymer. The small angle diffuse scattering is equivalent to the sum of scattering from many large, irregularly spaced particles. In synthetic fibres this diffuse scattering is due, primarily, to microvoids (Reference 35). As the scattering angle is inversely proportional to the correlating distance, an oriented specimen results in an elongation of the diffuse small angle scatter perpendicular to the orientation direction; in many spun synthetic fibres this effect is due mostly to elongation and orientation of the microvoids. One way of confirming that diffuse patterns are due to microvoids is to perform a series of SAXS experiments on swollen specimens. The physical structure of the fibers in terms of the percentage of crystallinity, crystal orientation in terms of crystallinity index (CI) and the orientation of the cellulose microfibrils in terms of the multifibrillar angle (MFA) were determined using two types of X-ray diffractometers. A Rigaku D-max/BH/2H X-ray diffractometer (Rigaku Americas, Woodlands, TX) with Bragg–Brentano parafocusing geometry, a diffracted beam monochromator, and a copper target X-ray tube set to 40 kV and 30 mA at a CuKa wave length of 1.5418 A ˚ was used to determine the percentage of 62 63 crystallinity, crystal size and CI. These measurements were taken on fibers that were made into pellets approximately 5 mm thick. To make the pellets, fibers were powdered in Wiley mill (Thomas Wiley, Swedesboro, NJ) to pass through a 250 lm mesh and the powdered fibers were pressed into a pellet using a hydraulic press operated at about 12,000 PSI. The percentage of crystallinity of the fiber was obtained by integrating the area under the crystalline peaks after subtracting the background and air scatter. CI was calculated as the ratio of the intensity differences in the peak positions at 18º and 22º according to Eq. (1) given below (Hindeleh, 1980; Mwaikambo and Ansell, 2002) where I22 and I18 are the intensity counts corresponding to a 2θ of 22º and 18º, respectively. A Bruker D8 Discover (Bruker AXS Inc., Madison, WI) model diffractometer equipped with an area detector and GAADS software was used to calculate the orientation of the cellulose microfibrils in the fiber to the fiber axis in terms of MFA and also to observe the diffraction pattern of the cellulose crystals in the fiber. The diffraction patterns were collected by mounting a bundle of fibers vertically in a specially designed sample holder. The diffraction patterns were collected for 10 min with the X-ray beam set to 40 kV and 30 mA. The 002 peak intensities in the diffraction patterns were fit into two Gaussian curves using a non-linear least square algorithm with the software 3.6 Nuclear magnetic resonance In the presence of a magnetic field, nuclei with non-zero nuclear spin have a discrete number of possible energy levels. Application of a radiofrequency pulse of the correct frequency can stimulate the nuclei into a higher energy state - the resonance condition. Nuclei with even atomic number, Z, and even atomic weight, A, possess zero spin; if A is even and Z odd the spin is an integer value; all other conditions result in half-integer values of spin. The spin is measured in units of h/2 where h is Planck's constant. The spin is characterised by the spin quantum number I = 1/2, 1, /2, 2, 5/2, ...etc. Thus H, C and F all have I = 1/2, Li, D and N have I = 1, and Na and Cl have I = 3/2 . C and O both have zero spin. The number of energy levels is (2I+1), thus nuclei of spin 1/2 have two possible energy levels and align either parallel or anti-parallel to the applied magnetic field; I = 1 results in three possible energy levels. The magnetic moment, magnetic moment is given by: , precesses about the direction of the magnetic field. The where is a characteristic of the nuclei called the gyromagnetic ratio. The precession (Larmor) frequency, , of the magnetic moment is given by: where is the applied magnetic field, is called the nuclear g factor, and is the nuclear magneton. The possible energy levels accessible to the nucleus in a magnetic field are characterised by a 63 64 magnetic quantum number m which has values I, (I - 1), (I - 2),......, -I. The potential energy, E, associated with each energy level is : and the energy difference is: Application of an RF field will stimulate transitions between energy levels if it is perpendicular to the applied steady field, , and if its frequency, , satisfies the resonance condition: The resonance frequency in a 1T field is 42.576 MHz for H and 10.705MHz for C. 3.7 Rheological properties Rheology is the science of the deformation and flow of matter, which is concerned with the materials on applied stress relating to irreversible viscous flow, reversible elastic deformation, or a combination of the two. The control of the rheology is basically for manufacture and handling of numerous materials and products such as the foods, cosmetics, rubber, plastics, paints, inks, and drilling mud. Deformation is a relative displacement of points of a body and could be understood with two types, e.g. flow and elasticity. In principal, flow is an irreversible deformation when the stress is removed, while the material does not revert to its original form indicating the work is converted to heat, and elasticity is a reversible deformation relating to the deformed body recovers its original shape, when the applied work largely recoverable. The viscoelastic materials are known to have both the flow and elasticity. Monsanto capillary rheometer 64 65 Monsanto Rheometer 100 65 66 3.7.1 Viscosity As a material, liquid continues to deform as long as it is subjected to a tensile and/or shear stress. The latter is a force applied tangentially to the material. For a liquid, the shear stress produces a sliding of one infinitesimal layer over another, that results in a stack-of-cards type of flow as Figure 3.7.1a shown. For a liquid under shear, the deformation rate or shear rate is a function of the shearing stress. The original exposition of this relationship is the Newton’s law, which defines a ratio of the stress to the shear rate in constant regarding as the viscosity. Under this condition, viscosity is independent of shear rate, and such liquids thus been regarded as the Newtonian liquids. However, the viscosities of most liquids, especially those broadly appeared in industry are not independent of the shear rate and been regarded as the non-Newtonian liquids. Figure 3.7.1a. Laminar flow in simple shear. F/A = η dV/dX, where F is the force acting on area A, V the velocity and X the thickness of the layer, and η the coefficient of viscosity or the Newtonian viscosity. Some commonly types of flow behavior are described in Figure 3.7.1b by taking the shear stress as a function of the shear rate and this plot is usually called the flow curves for understanding the rheological behavior of liquids. Of which, the Newtonian flow is described by a straight line, and shear thinning and thickening by curves. Yield stresses, τ0, are shown by intercepts on the stress (y) axis. Figure 3.7.1b. A typical flow curve for different types of flow behavior. Viscosity, η, is equal to the slope of the flow curve, dτ/dγ . The quantity τ/γ is the viscosity η for a Newtonian liquid and the apparent viscosity ηa for a non-Newtonian liquid. The kinematic 66 67 viscosity is the viscosity coefficient divided by the density, ν = η/ρ. The fluidity is the reciprocal of the viscosity, φ = 1/η. The common units for viscosity, dyne seconds per square centimeter ((dyn·s)/cm2) or grams per centimeter second ((g/(cm·s)), called poise, which is usually expressed centipoise (cP), have been replaced by the SI units of pascal seconds, e.g. Pa·s and mPa·s, where 1 mPa = 1 cP. In same places, the shear stress units of dynes per square centimeter, dyn/cm2, has been also replaced by Pascals, 10 dyn/cm2=1 Pa, and 1 N/m2=1 Pa. The shear rate is V/X, or length/time/length, its value is presented as, s−1. The SI units for kinematic viscosity are square centimeters per second (cm2/s), and square millimeters per second (mm2/s). Different flow models are summarized in Table 3.7.1a. Table 3.7.1a. Different flow models. The power law, τ =kγn, is widely used as a model for defining the non-Newtonian fluids. It holds for many solutions and can describe Newtonian, shear-thinning, and shear thickening behavior, depending on the power factor, n, also called the flow behavior index. For a Newtonian fluid, n = 1 and the equation reduces to the Newtonian model. If n is less than 1, the fluid is shear thinning; if it is greater than 1, the fluid is shear thickening. A test of whether the power law applies and a means of determining n is to plot the log shear stress vs the log shear rate. If the plot is linear, the power law applies. The value of n, which is the reciprocal of the slope of the line, can be used as a measure of the degree of shear thinning or shear thickening. Dividing the power law equation through by γ gives an expression in terms of viscosity, η = k’γn−1. The power law model can be extended by including the yield value τ−τ0=kγn, which is called the Herschel–Bulkley model, or by adding the Newtonian limiting viscosity η∞. The latter is done in the Sisko model, η∞+kγn−1. These two models, along with the Newtonian, Bingham, and Casson models, are often included in data-fitting software supplied for the newer computer-driven viscometers. Another model is the Casson equation (11), which is useful in establishing the flow characteristics of inks, paints, and other dispersions. An early form of this expression (eq. 1) was modified (12) to give equation 2. τ 1/2 =k0 +k1 γ1/2 () 1/2 1/2 1/2 −1/2 η =η∞ +τ0 γ () 67 68 Figure 3.7.1c. The Casson plots. The Williamson equation is an useful model for describing the relationship between the shear-thinning fluids over a wide range of shear rates (Eq. ). Of which, τ is the absolute value of the shear stress and τm is the shear stress at which the viscosity is the mean of the viscosity limits η0 and η∞, i.e. at η = (η0 + η)∞/2. The Cross equation indicated that a shear-thinning fluid has high and low shear-limiting viscosity (Eq. ), where α and n are two constants. The n is often valued as 2/3, but for most polymer melts this value is varied with a wide range. The constant α is associated with rupture of the linkages in the structure of the fluid. The effect of different values of α, ie, at the same values of η0 and η∞, is shown in Figure 4. As α increases, breakdown occurs at lower and lower shear rates. 68 69 Figure 3.7.1d. Plots of viscosity vs shear rate corresponding to the Cross model relating to different α values. Monsanto Mooney viscometer 3.7.2 specific viscosity The viscosity ratio or relative viscosity, ηrel, is the ratio of the viscosity of the polymer solution to the viscosity of the pure solvent. In capillary viscometer measurements, the relative viscosity (dimensionless) is the ratio of the flow time for the solution t to the flow time for the solvent t0 (Table 2). The specific (sp) viscosity (dimensionless) is also defined in Table 2, as is the viscosity number or reduced (red) viscosity, which has the units of cubic meters per kilogram (m3/kg) or deciliters per gram (dL/g). The logarithmic viscosity number or inherent (inh) viscosity likewise has the units m3/kg or dL/g. For ηred and ηinh, c, the concentration of polymer, is expressed in convenient units, traditionally g/100 cm3 but kg/m3 in SI units. The viscosity number and logarithmic viscosity number vary with concentration, but each can be extrapolated (Fig. 8) to zero concentration to give the limiting viscosity number (intrinsic viscosity) (Table 2). Usually, measurements at four or five concentrations are needed. The specific viscosity is represented by Eq. ? 69 70 Figure 3.7.1e. Plots of viscosity number (ηrel = ηsp/c) and the logarithmic viscosity number (ηinh = ηrel/c) vs concentration. Extrapolations to zero concentration give the limiting viscosity number [η]. where kh is the Huggins viscosity constant, which was usually applied to describe dilute solution viscosity number or index and could be easily determined from the slope of a plot of ηsp/c vs c such as the lower plot in Figure 3.7.1e. The Huggins constant can be thought of as a measure of the “goodness” of the solvent for the polymer with values around 0.3 in good solvents and 0.5–1 in theta solvents and it can be found in Reference 25 along with constants for another semi-empirical equation relating viscosity and concentration, that of Schulz and Blaschke (27). Table 3.7. The Mark–Houwink equation described a new relationship between the viscosity and molecular weight of polymer, [η]=KMa, where K and a are two constants and M is the 70 71 viscosity-based molecular weight of the polymer. Values of K and a are varied dependence of polymers and solvents (31,32), and they may be influenced by several factors, foe example, the K constant has been found strongly in relation to the polarity of polymer (Shen et al. JCIS 2004). Ray Ran melt flow indexer 3.5 Bio-properties 3.5.1 Anti-bacteria 71 72 3.5.2 3.6 Nano properties 3.7 Other properties Benz abrasion tester Branson sonic welder 72 73 Carver press 1 73 74 Carver press 2 compression press 1 74 75 compression press 2 curing oven 75 76 Denver Instruments, moisture analyzer Elmer-Perkin infrared spectrophotometer Recommending reading 76 77 4 Typical materials and related fiber spinning techniques 4.1 Synthetic polymer-based fiber spinning 4.1.1 Polyamide (PA) PA6 PA66 Figure 4.1.2 Polyacrylonitrile (PAN) Figure 4.1.3 Polypropylene (PP) Figure 4.1.4 Polyester (PET,PBT,PTT) PET 77 78 PBT Figure 4.1.5 Polyethylene (PE) Figure K=2 M = 42; DP > 62,500 rM=107...138°C TG^ - 1000C (-20%-300C) 4.1.6 Polyurethanes (PU) 4.1.7 Polyvinyl chloride (PVC) Figure 4.1.8 Polyvinyl Alcohol (PVA) 4.1.9 Polytetrafluorethylene Fibers (PTFE) 4.1.10 Polylactic acid (PLA) Cargill Inc. has been the company most active in the development of poly(lactic acid) 78 79 (polylactide, PLA). After nine years of lactic acid and poly(lactic acid) development, Cargill Dow polymers LLC was created in 1997 as a joint venture of Cargill and the Dow Chemical company. Figure The isomers of lactic acid. Figure 79 80 4.2 Natural polymer-based fiber spinning 4.2.1 Cellulose Cellulose is the most abundant natural polymer, and is the structural component of plant cell walls. It is a linear polysaccharide consisting D-anhydroglucopyranose units (often abbreviated as anhydroglucose units or as glucose units for convenience) linked together by b-(1 ! 4)-glycosidic bonds, as shown in Fig. ?. 4.2.1.1 Viscose fiber 4.2.1.2 Lyocell fiber 4.2.1.3 Others 4.2.2 Chitin Chitin is the second most abundant natural polymer (after cellulose) on earth. It is a linear polysaccharide of b-(1-4)-2-acetamido-2-deoxy-D-glucose. The chemical structure of chitin is very similar to that of cellulose with a hydroxyl group replaced by an acetamido group. Pure chitin with 100% acetylation does not exist in nature. Chitin tends to form a co-polymer with its N-deacetylated derivative, chitosan. Chitosan is a polymer of b-(1-4)-2-amino-2-deoxy-D-glucose. The chemical structures of cellulose, chitin and chitosan are shown in Fig. 27 [84]. When the fraction of acetamido groups is more than 50% (more commonly 70–90%), the co-polymer is termed chitin. The co-polymer consists of chitin and chitosan units randomly or block distributed through out the polymer chain, as shown in Fig. 28 [85]. Chitin is widely distributed in fungal and yeast cell walls, mollusk shells, arthropod exoskeletons and other invertebrates. It plays an important role as the structural component that provides support and protection to the organisms. 80 81 Figure 4.2.3 Protein 4.2.4 Lignin 4.3 Inorganic fiber 4.3.1 Carbon fiber The carbon fiber is valuable on the basis of its unique combination of extremely high modulus and strength and low specific gravity. The precursors of carbon fiber have been known usually the pitch, rayon, or polyacrylonitrile, PAN, fiber. Of which the rayon and pitch fibers are less employed as precursor. The precursors converted into the carbon fibers usually required two-stage thermal treatments, a middle temperature-based oxidation stage and a high temperature-based carbonization. After these two stages, the fiber could be converted into nearly pure carbon. 4.3.2 Oxide fiber 4.3.3 Silicon fiber 81 82 4.3.4 SiC fiber 4.3.5 Glass fiber primarily low alkali, Bor silicate glass, e.g.,about 52% SiO2, about 11% B2O3, about 14% AI3O3 and about 17% CaO. Recommending reading [1] M. G.Northolt, P. den Decker, S. J. Picken, J. J. M. Baltussen, R. Schlatmann, The Tensile Strength of Polymer Fibres, Adv Polym Sci (2005) 178: 1–108 [ ] Toshihiro Ishikawa, Advances in Inorganic Fibers, Adv Polym Sci (2005) 178: 109–144 1 The collection of fiber products 1.1 characteristics and industrial uses Generic Fibers Characteristics Major Domestic and Industrial Uses ACETATE Luxurious feel and appearance Apparel: Blouses, Wide range of colors and lusters garments. Excellent drapability lingerie, dresses, linings, and foundation shirts, slacks, and sportswear. softness Fabrics: Brocade, crepe, double knits, faille, Relatively fast-drying knitted jerseys, lace, satin, taffeta, tricot. Shrink-, moth-, and Home Furnishings: Draperies, upholstery. mildew-resistant Other: Cigarette filters, fiberfill for pillows, quilted products 82 83 ACRYLIC Soft and warm Apparel: Dresses, infant wear, knitted garments, Wool-like ski wear, socks, sportswear, sweaters. Retains shape Fabrics: Fleece and pile fabrics, face fabrics in Resilient bonded fabrics, simulated furs., jerseys. Quick-drying Home Furnishings: Blankets. carpets, draperies, Resistant to moths, sunlight, oil upholstery. and chemicals Other: Auto tops, awnings, hand-knitting and craft yarns, industrial and geotextile fabrics. ARAMID Does not melt Hot-gas filtration fabrics, protective clothing, Highly flame-resistant military helmets, protective vests, structural High strength composites for aircraft and boats, sailcloth, tires, High resistance to stretch ropes and cables, mechanical rubber goods, Maintains its shape and form at marine and sporting goods. high temperatures LYOCELL Soft, strong, absorbent Dresses, slacks and coats. Good dyeability Fibrillates during wet processing to produce special textures MELAMINE White and dyeable Flame resistance Fire Blocking Fabrics: Aircraft seating, fire and low blockers for upholstered furniture in high-risk thermal conductivity occupancies (e.g., to meet California TB 133 High heat dimensional stability requirements) Processable on standard textile Protective Clothing: Firefighters' turnout gear, equipment insulating thermal liners, knit hoods, molten metal splash apparel, heat resistant gloves. Filter Media: High capacity, high efficiency, high temperature baghouse air filters. MODACRYLIC Soft Apparel: Deep pile coats, trims, linings, simulated Resilient fur, wigs and hairpieces. Abrasion- and flame-resistant Fabrics: Fleece fabrics, industrial fabrics, knit-pile Quick-drying fabric backings, non-woven fabrics. Resists acids and alkalies Home Furnishings: Awnings, blankets. Carpets, Retains shape flame-resistant draperies and curtains, scatter rugs. Other: Filters, paint rollers, stuffed toys. 83 84 NYLON Exceptionally strong Apparel: Blouses, dresses, foundation garments, Supple hosiery, lingerie and underwear, raincoats, ski Abrasion-resistant and snow apparel, suits, windbreakers. Lustrous Home Easy to wash draperies, curtains, upholstery. Furnishings: Bedspreads, carpets, Resists damage from oil and Other: Air hoses, conveyor and seat belts, OLEFIN many chemicals parachutes, racket strings, ropes and nets, Resilient sleeping bags, tarpaulins, tents, thread, tire cord, Low in moisture absorbency geotextiles. Unique wicking properties that Apparel: Pantyhose, underwear, knitted sports make it very comfortable shirts, men's half hose, men's knitted sportswear, Abrasion-resistant sweaters. Quick-drying Home Furnishings: Carpet and carpet backing, Resistant to deterioration from slipcovers, upholstery. chemicals, mildew, perspiration, Other: Dye nets, filter fabrics, laundry and rot, and weather sandbags, geotextiles, automotive interiors, Sensitive to heat cordage, doll hair, industrial sewing thread. Soil resistant Strong; very lightweight Excellent colorfastness POLYESTER Strong Resistant Apparel: to stretching Blouses, shirts, career apparel, and children's wear, dresses, half hose, insulated shrinking garments, ties, lingerie and underwear, Resistant to most chemicals permanent press garments, slacks, suits. Quick-drying Home Furnishings: Carpets, curtains, draperies, Crisp and resilient when wet or sheets and pillow cases. dry Other: Fiberfill for various products, fire hose, Wrinkle- and abrasion-resistant power belting, ropes and nets, tire cord, sail, Retains heat-set pleats and V-belts. creases Easy to wash PBI Highly flame resistant Outstanding combined comfort with Suitable for high-performance protective factor apparel such as firemen's turnout coats, astronaut thermal and space suits and applications where fire resistance chemical stability properties is important. Will not burn or melt Low shrinkage, when exposed to flame. 84 85 RAYON Highly absorbent Apparel: Blouses, coats, dresses, jackets, lingerie, Soft and comfortable linings, millinery. rainwear, slacks, sports shirts, Easy to dye sportswear, suits, ties, work clothes. Versatile Home Furnishings: Bedspreads, blankets, carpets, Good drapability curtains, draperies, sheets, slipcovers, tablecloths, upholstery. Other: Industrial products, medical,: surgical products, non-woven products, tire cord. SPANDEX Can be stretched 500 percent Articles (where stretch is desired): Athletic without breaking apparel, bathing suits, delicate laces, foundation Can be stretched repeatedly garments, golf jackets, ski pants, slacks, support and recover original length and surgical hose. Light-weight Stronger and more durable than rubber Resistant to body oils Fiber type, trade names and manufacturers Fiber Type Trade Names Manufacturer ACETATE Celanese, MicroSafe Chromspun, Estron Celanese Acetate Eastman Chemical Company ACRYLIC Acrilan, Bounce-Back, Solutia Inc. Duraspun, Evolutia, Ginny, Pil-Trol, So-Lara, The Smart Yarns, Ware-Dated BioFresh, CFF Fibrillated Sterling Fibers, Inc Fiber, Conductrol, Creslan, Cresloft, MicroSupreme, WeatherBloc Kevlar, Nomex DuPont Company BICOMPONENT Resistat BASF Corporation FLUORO Teflon DuPont Company ARAMID 85 86 LYOCELL Fibro, Galaxy Lyocell by Lenzing Acordis Cellulosic Fibers, Inc. Lenzing Fibers Corporation MELAMINE Basofil BASF Corporation MODACRYLIC SEF Plus Solutia Inc. MODAL Micro-ProModal, MicroModal Modal by Lenzing, Modal Sun ProModal Lenzing Fibers Corporation NYLON 6 Anso, Anso AllSport, Anso Honeywell International Caress, Anso Choice!, Anso Color Solutions, Anso CrushResister III, Anso CrushResister III ACT, Anso CrushResister III Select, Anso CrushResister TLC, Anso HTX, Anso Premium, Anso Replacement Plus, Anso Soft Anso Solution, Anso Total Comfort, Anso Vibrance, Powersilk, Shimmereen Caprolan, Dry Step, Eclipse, BASF Corporation Hardline, Hydrofil, Matinesse Micro Touch, Nylon 6ix, Softglo, Sportouch, Stay Gard Tru-Ballistic, Ultra Touch, Zefsport, Zeftron 200, Zeftron 2000, Zeftron 2000ZX NYLON 6.6 Antron, Antron Advantage, DuPont Company Antron II, Antron Legacy, Antron Lumena, Assurance, Avantige, Cantrece, Comforlast, Cordura, Hytel, Micro Supplex, Natrelle BCF, No Shock, OPTA, Stainmaster Stainmaster Luxra, Stainmaster XTRA Life, Supplex, Tactel, Tactel Aquator, Tactel Ispira, Tactel Strata Durasoft, Duratrek, DyeNAMIX, Tactel Micro, Solutia Inc. Traffic Control Fiber System 86 87 Ultramirage Ultron Ultron 3D Ultron VIP Wear-Dated Wear-Dated Assurance Wear-Dated Freedom Wear-Dated II Enka Nylon Acordis Industrial Fibers Inc. NYLON 6 or 6.6 Wellon, Wellstrand Wellman, Inc. OLEFIN Alpha BCF, Essera, Impressa Innova, Marqesa Lana, Trace Trace FR Elustra, Herculon, HY – Colour, HY – Comfort, HY – Medical, HY – Repeat, Nouvelle, Soft 71 Permafresh Spectra 1000, Spectra 900, Spectra Fusion, Spectra Guard Spectra Shield, Spectra Shield Plus, SpectraFlex American Fibers and Yarns Company PBI PBI Logo Celanese Acetate A.G. PELCO Securus Honeywell International PEN Pentex Honeywell International POLYESTER A.C.E. Polyester, DSP, Fiberbrite 2000, Securus, Stay Gard, Substraight Avora FR, Celbond, Easy Dye Fibers, ESP, Loftguard Loftguard Xtra, Microtherm, Polyguard 3D, Polyguard Classic, Polarguard Home, Polyguard HV, Serelle, Serene Colorfine, Nature Tex Comforel, CoolMax, CoolMax Alta, Hollofil, Microloft, MicroMattique, Honeywell International 87 FiberVisions Incorporated Drake Extrusion Honeywell International KoSa Martin Color-Fi, Inc. DuPont Company 88 Microselect, Quallofil, Qualloform Corebond, Fillwell, Fillwell II, Fillwell Plus, Fortrel, Fortrel BactiShield, Fortrel EcoSpun Fortrel EcoSpun2, Fortrel MicroSpun, Fortrel Plus, Fortrel Spunnaire, Fortrel Spunnese, Sensura, Wellene Dacron Diolen Tairilin Wellman, Inc. DuPont-Akra Polyester, LLC Acordis Industrial Fibers, Inc. Nan Ya Plastics Corporation, America RAYON Fibro, Galaxy Acordis Cellulosic Fibers, Inc. Viscose by Lenzing , Viscose Lenzing Fibers Corporation FR by Lenzing SPANDEX Cleerspan, DC-100, DC-700, Globe Manufacturing Company Glospan Dorlastan Bayer Lycra DuPont Company 88 89 89 90 90 91 91 92 92 93 93 94 94 95 95 96 96 97 97 98 98 99 99 100 100 101 101 102 102 103 103 104 104 105 105 106 106 107 107 108 108 109 109 110 110 111 111 112 112 113 113 114 114 115 115 116 116 117 117 118 118 119 119 120 120 121 Table ? Typical apparel fiber blend ratios. 121