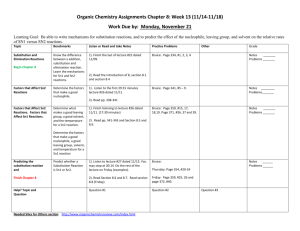
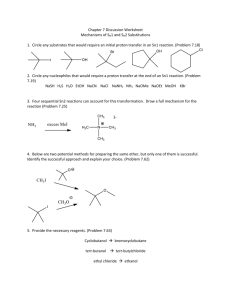
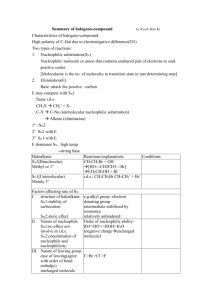
Chapter 11: Ionic Substitution Reactions
advertisement

Chapter 11: Ionic Substitution Reactions Note to students: This is a single chapter from a textbook that is under construction. Therefore you can ignore references to other textbook sections. 11.1 Why Should I Study This? In a substitution reaction, one portion of the molecule is replaced by something new. In most organic substitution reactions, the new piece that adds to the molecule, or the old piece that is replaced, or both, are ions. Therefore this important process is frequently referred to as an ionic substitution reaction. Ionic substitution at sp3 carbon (the topic of this chapter) is also called nucleophilic aliphatic substitution. Ionic substitution is among the simplest and most fundamental, as well as most common organic reactions. It usually results in the conversion of one functional group, such as an alkyl halide, into a new functional group. Replacement of one functional group with another is a fundamental operation in organic synthesis, the science (and art!) of converting simple organic molecules into more complex molecules such as pharmaceuticals. (We study organic synthesis in chapters 14 and 33.) Reaction 11.1 illustrates an ionic substitution reaction in which an alkyl chloride is converted into a thiol. This reaction is the last step in the manufacture of captopril, an antihypertensive drug that works by inhibiting angiotension converting enzyme. Alkyl chloride Thiol CH3 CH3 Cl NaSH N HS DMF, 4 hr, 50o CO2H O 11.1 N CO2H O Captopril Ionic substitution is also important in numerous biochemical processes. For example, S-adenosylmethionine (SAM) can add a methyl group to another molecule to meet a variety of cellular needs. SAM is the focus of this chapter’s In the Real World discussion, section 11.10. NH2 This methyl group is transferred N O N CH3 S N O O NH3 HO OH S-adenosylmethionine (SAM) Ionic Substitution Reactions – Page 1 N While exploring ionic substitution reactions we will also become familiar with some basic features that are common to many other organic reactions: nucleophiles, leaving groups, steric effects and solvents. 11.2 The SN2 Reaction Review Conjugate base (9.xx), curved arrows (3.xx) and reaction mechanism (3.xx). Let’s begin our study of ionic substitution reactions by revisiting a substitution reaction that we are already familiar with: an acid-base reaction. In an acid-base reaction, a base (often an ion) attacks a hydrogen atom of an acid, causing the conjugate base to leave. In other words, the base replaces the conjugate acid. For example, hydroxide ion reacts with hydrogen chloride to produce water and chloride ion (reaction 11.2). Note the curved arrows that show the bond changes and summarize the reaction mechanism. HO H Cl δ+ δ- HO H + 11.2 Cl You can imagine that reaction 11.2 is initiated by the attraction of opposite charges: the negative formal charge of HO- and the δ+ of HCl. This electrostatic attraction is a common feature of ionic substitution reactions, and many other organic reactions in general. (Flip through this text and you will find many examples.) In Chapter 9 we learned quite a bit about acid/base reactions and what influences them. Now it’s time to extend what we have learned to a new and very important class of reactions. Think Ahead Question 11.1 Based on reaction 11.2, write the reaction that might occur between HO- and CH3Cl. Include all curved arrows, formal charges and lone pairs. Will this reaction occur as written? Answer: Changing HCl to CH3Cl gives us reaction 11.3. H H HO H C + Cl δ δH HO C H + Cl 11.3 H Will this reaction occur as written? Like the HO-/HCl reaction (11.2), we can also imagine this reaction is initiated by interaction of opposite charges. In this case the positive charge is the δ+ carbon atom of CH3Cl. So if our charge interaction model is valid, reaction 11.2 will occur as written. It is useful at this point to review some terms we learned previously that also apply to this substitution reaction (as well as many other reactions we will encounter throughout our studies of organic chemistry). Ionic Substitution Reactions – Page 2 Nucleophile: The reactant that provides the electron pair to form a new covalent bond. Also called a Lewis base. Nucleophiles usually have lone pairs, pi bonds, formal negative charges, or (less commonly) δ- charges. Electrophile: The reactant that accepts the pair of electrons to form a new covalent bond. Also called a Lewis acid. Electrophiles usually have open octets, formal positive charges or δ + charges. Some electrophiles also have a leaving group as part of their structure. Leaving group: An atom or group that breaks away, taking with it the electron pair that used to form the covalent bond between the leaving group and the rest of the molecule. Nucleophile Electrophile Leaving group H H HO H HO C + Cl δ δ− H C H + Cl H Why is this reaction interesting? We start with methyl chloride (an alkyl halide) and convert it into methanol (an alcohol). We have achieved a functional group conversion. As we learned in section 11.1, functional group conversions are important in a variety of organic reactions, so this ionic substitution is worthy of further scrutiny. In the coming sections we will explore certain features of this simple reaction that apply to other substitution reactions and many other types of more complex organic reactions as well. In general the single step ionic substitution mechanism can be drawn this way, where Nuc = nucleophile and LG = leaving group: R1 Nuc R1 C LG Nuc + LG C R2 R2 R3 R3 Concept Focus Question 11.1 Write the products and mechanisms for the following ionic substitution reactions. Include all formal charges and lone pairs. Label each nucleophile, electrophile and leaving group. Describe the functional group change. CH3 (a) CH3Cl + I (c) H C CH3 (b) CH3O + CH3Cl O Cl + C O (d) CH3 + C N Br Concept Focus Question 11.2 Write the missing reactants on the left side of the arrow as necessary to complete each reaction. Include all formal charges, lone pairs and curved arrows. Label Ionic Substitution Reactions – Page 3 Review Nucleophile, electrophile and leaving group (8.xx). Despite its name, a leaving group is not always a group of atoms. I-, Br-, Cl- and F- are called leaving groups even though they are single atoms. each nucleophile, electrophile and leaving group. Describe the functional group change. (a) CH3Br + ??? (b) ??? + SCH3 CH3I CH3CH2SCH3 (c) CH3CH2I + ??? (d) ??? + OC(CH3)3 CH3CH2NH3 I OC(CH3)3 The reaction of an alkoxide ion (RO-) with an alkyl halide or alkyl sulfonate is called the Williamson ether synthesis, and is discussed in more detail in section 20.xx. Concept Focus Question 11.3 What nucleophile is necessary to convert CH3Cl into each of the following? (a) CH3I (e) CH3SC6H5 (i) CH3NH3+ Cl(b) CH3OH (f) CH3C≡N (j) (CH3)4N+ Cl(c) CH3OCH3 (g) CH3C≡CH (k) CH3OH2+ Cl(d) CH3SH (h) CH3N3 (l) C6H5CO2CH3 A. Mechanism and Kinetics In order to make any practical use of the ionic substitution reaction, we must understand how various factors influence its rate. For example, if we use Iinstead of Br- will the reaction be faster or slower, or perhaps so slow that the product will not be produced at a measurable rate? (The rate issue is of very broad concern. For example, in the chemical industry, time is money, and a reaction that takes too long is of no value.) Review Connection between reaction rate and mechanism (8.xx). Studying the reaction rate is also a useful window on the reaction mechanism. We can write a mechanism on paper, but how do we know that this is what the reactants are actually doing? We proposed a mechanism in the previous section: the two bond changes occur simultaneously. In other words the reaction is concerted. Review if necessary the relationship between reaction rate and mechanism (section 8.xx). Think Ahead Question 11.2 Write the reaction rate expression for reaction 11.3. Answer: Recall from section 8.xx (and your studies of reaction kinetics in introductory chemistry) that for a reaction in which all bond changes occur in a single step, the rate expression is the product of the concentration of each reactant. The rate expression is first-order in each reactant and second-order overall: Rate = k [CH3Cl][HO-] This rate expression tells us that changing the concentration of either CH3Cl or HO- changes the rate of the reaction. For example, doubling the concentration of CH3Cl doubles the rate. Reducing the concentration of HO- to 1/10 of the starting value cuts the reaction rate by ten. Ionic Substitution Reactions – Page 4 The rate expression is meaningful only if the mechanism is accurate. Conversely, if the mechanism prediction is inaccurate then the effects on reaction rate caused by changing concentration of reactants will not agree with the rate expression. (We are assuming that the reaction does not have a more complex mechanism whose observed rate expression is much simpler than the mechanism suggests. There are other ways to test for this in the lab.) In the lab we find that the predicted rate expression agrees with experiment, which suggests that the mechanism is reasonable. (A rate study can disprove a predicted mechanism, but it can never prove the mechanism because we can write other mechanisms whose rate expressions will appear to be identical.) In fact, lab studies have shown that many simple ionic substitution reactions follow this same rate law, which we can write in a generic form: Rate = k [nucleophile][electrophile] Ionic substitution reactions that follow this rate law are called SN2 , an abbreviation for substitution nucleophilic bimolecular. (Bimolecular means the rate law is first-order in two reactants.) In modern sense, any ionic substitution process in which the nucleophile bonds to carbon at the same time the leaving group leaves is called SN2, even if it is not bimolecular. For example, the nucleophile and leaving group might be contained within the same molecule, resulting in an intramolecular substitution reaction and a first-order rate expression (reaction 11.4). We still call this an SN2 reaction. O O O 11.4 Rate = k I I The term SN2 was first introduced by Edward Hughes and Sir Christopher Ingold of the University College in London, who carried out very extensive studies on ionic substitution reactions beginning about 1930. They preformed early studies in physical organic chemistry, especially in substitution and elimination reactions. They introduced many concepts and terms in wide use today such as nucleophile, electrophile, inductive effect, SN2 and SN1. Ingold was the co-author of the Cahn-Ingold-Prelog priority rules (section 6.xx). Reaction 11.4 may appear incomplete because it does not include the leaving group (I-) that has departed or the cation (such as Li+ or Na+) that is associated with the oxyanion (RO-). This is normal. These inorganic species are of little interest to us (we want to synthesize more complex organic molecules, not make simple metal salts), so we often leave them out when we write reactions and mechanisms. Ionic Substitution Reactions – Page 5 Caution! In a significant number of organic reaction mechanisms, the leaving group still participates after it has left. It is not necessary to draw it in the mechanism step in which it has left, but do not forget about its presence. If you are the forgetful type (even slightly so), it might be a good habit to draw the departed leaving group just to remind yourself. On the other hand, metal cations rarely play an important role in the reaction mechanisms we will study, so they can usually be ignored without significant consequences. Concept Focus Question 11.4 Each of the following is an SN2 reaction. Write the product(s) (including departed leaving group), mechanism and rate law. If you are having trouble with the products and mechanism, look for patterns from previous examples, such as the kinds of things that can be leaving groups or nucleophiles. (a) (b) Na I Br S Li (c) CH3I I H2O (d) O Cl B. Transition State Transition state and energy of activation (8.xx). When we studied the fundamental concepts of reactions in Chapter 8, we learned that the structure of the transition state and the energy of activation influence the reaction rate. Altering any factor in the reaction that changes the stability (energy) of the transition state and/or reactants can change the reaction rate. Therefore if we are interested in understanding how various factors influence the rate of an SN2 reaction, we must pay attention to the transition state. transition state This energy difference controls rate energy Review Reactants Products reaction coordinate Figure 11.1: Energy diagram for a typical exergonic SN2 reaction. Ionic Substitution Reactions – Page 6 Think Ahead Question 11.3 Draw the structure of the transition state for reaction 11.3. Answer: Even though a transition state has a very brief existence – no more than the time it takes for one bond vibration to occur – it still obeys the same rules and forces as any other molecular structure. The most broadly influential of these forces is electron repulsion, which we explored in section 2.xx. Just to review, the attachments (atoms, groups of atoms and lone pairs) arrange themselves around a central atom in a way that minimizes electron repulsion. Size matters: larger groups cause more electron repulsion, causing smaller groups to move away a little bit. The position of the attachments around the central atom controls its hybridization. If the central atom has four attachments, a tetrahedral arrangement gives the least electron repulsion and the lowest energy, and the central atom uses sp3 hybrid orbitals to bond with the attachments. The central atom of our transition state (a carbon) has five attachments. These are best accommodated using a trigonal bipyramid: two pyramids with triangular faces and a common base. So far our transition state looks like this: C The nucleophile (HO-) is approaching the carbon in order form a bond, but that bond is not yet complete. We indicate this by using a dashed line that is longer than a normal (full) bond. The nucleophile is using a lone pair to make its new bond to carbon, so its electron density (as measured by formal charge) is decreasing. In HO- the oxygen bears a formal negative charge and in the product it is neutral. In the transition state its charge is not quite -1 but not yet zero, so we indicate this partial negative charge using our familiar δ- symbol. The leaving group (Cl-) is on its way out (along with the electron pair that was the C-Cl bond) but not yet completely gone. Like the partial carbon-nucleophile bond, we use an elongated dashed line to depict the partial carbon-leaving group bond. Like the nucleophile, its charge is changing (neutral in CH3Cl to negative in Cl-) so in this transitory structure its charge is also transitory (δ-). Where should we place the nucleophile and leaving group? They both have a δcharge. To maximum stability (and minimize electron repulsion), we place the nucleophile and leaving group as far apart as possible. This arrangement in which the nucleophile approaches the carbon-leaving group bond from the carbon end is called backside attack. δHO C δCl The hydrogen atoms of the methyl group move around as the reaction occurs, but the C-H bonds do not break. They lie in the space somewhere between the Ionic Substitution Reactions – Page 7 Review Electron repulsion effects on molecular structure (2.xx). leaving group and nucleophile. We cannot predict exactly where they are (as discussed in section 8.xx), so we draw them about halfway between the nucleophile and leaving group. Don’t forget the wedge and dashed line, to show the hydrogens are coming out of and going into the plane of the paper. This reminds us of the three-dimensional shape of the structure. Lastly, we enclose the structure in brackets and add the little double dagger symbol (‡) indicate this is a transition state. The completed transition state drawing is shown in Figure 11.2. H δHO δCl C H H Simplified drawing Calculated Figure 11.2: Transition state representations for the SN2 reaction of HO- with CH3Cl. This drawing is just a crude sketch to give us an approximation of the relative position of each atom or group in the transition state, but it is good enough for our simple purposes. The actual transition state for this reaction, using molecular modeling software, is also shown in Figure 11.2. Build a model of the transition state to get a better feeling for the three-dimensional relationship of the atoms. Concept Focus Question 11.5 What is the formal charge or δ charge on the carbon atom in the transition state of Figure 11.2? Concept Focus Question 11.6 Carbon is limited to eight valence shell electrons (so it can form four covalent bonds at most) yet the carbon of the transition state in Figure 11.2 has five bonds. Explain. Concept Focus Question 11.7 Students often ask if it is necessary to draw a transition state in a certain way. For example, does the nucleophile have to be on the right, or does one C-H bond always have to be vertical? The transition state can be drawn from any viewpoint or orientation, as long as it accurately depicts the relative positions of the atoms involved. To explore this point, build a model of transition state A, then use it decide if structures B-D depict the same transition state. Cl δ- I δ- H δI δCl C H H3C CH2CH3 A H3C H δCl C CH2CH3 B δCl δI C CH3CH2 CH3 C Ionic Substitution Reactions – Page 8 CH2CH3 H C CH3 - Iδ D Concept Focus Question 11.8 (a) According to the Hammond postulate (section 8.xx), the structure of the transition state is influenced by the energy of activation. Draw the transition state for an exergonic SN2 reaction with a generic nucleophile (nuc) at a methyl group bearing a generic leaving group (CH3-LG). (b) Draw the transition state for an endergonic SN2 reaction with a generic nucleophile with CH3-LG. (c) What is necessary for the three hydrogens of these generic transition states to be exactly coplanar? Concept Focus Question 11.9 Draw the transition states for the reactions in Concept Focus Question 11.4. Assume each reaction is exergonic. C. Inversion of Configuration In the previous section we developed a transition state structure for the SN2 reaction of HO- and CH3Cl. We assumed the nucleophile would attack the backside of the carbon-leaving group bond, because the nucleophile and leaving group δ- charges repel. Can we verify this assumption experimentally? What is the experimentally observable difference (if any) between backside attack and front side attack (attack on the same face as the leaving group)? Think Ahead Question 11.4 Consider the SN2 reaction of (S)-2-iodobutane with cyanide ion (reaction 11.5). The product has a stereocenter so two enantiomers are possible (review enantiomers in section 6.xx). Based on the mechanism and transition state, predict which enantiomer(s) are produced. Models of the starting iodide and the transition state are very useful here. CH3CH2 CH3CH2 H C CH3 I C N H CH2CH3 C CH3 CN + NC H 11.5 CH3 Answer: As cyanide approaches the backside of the carbon-iodine bond, the ethyl group, methyl group and hydrogen atom begin to move away. (When someone crowds you on a bench, don’t you move away, too?) As the carbon bearing the leaving group changes from tetrahedral (sp3) in 2-iodobutane to trigonal planar (sp2) in the transition state, the attachments move from a tetrahedral to a planar arrangement. (The partial NC---C and C---I bonds lie along the sp2 carbon’s unhybridized p orbital.) As the leaving group departs, more space is made available. To reduce repulsion the attachments move away from the incoming nucleophile toward the newly vacated space, and the carbon returns to sp3. Ionic Substitution Reactions – Page 9 Review Enantiomers (6.xx). CH3CH2 CH3CH2 NC H C I δNC C CH2CH3 δI NC H CH3 H + I CH3 CH3 Reactants Transition state Products Figure 11.3: Atomic motions in the SN2 reaction leading to inversion of configuration. The arrows indicate direction of motion. Review Stereochemical configuration (6.xx). This model predicts that the reaction produces (R)-2-methybutyronitrile, a prediction that is consistent with experimental results. In fact studies of many SN2 reactions have shown that backside attack always occurs. Even if the nucleophile and leaving group have opposite charges in the transition state, backside attack still occurs, even though we might expect front side attack because opposite charges attract. (Explore this more in Concept Focus Question 11.14.) This backside attack leads to inversion of configuration at the asymmetric carbon. (Review the concept of stereochemical configuration in section 6.xx.) If the configuration does not change we call it retention of configuration.) Caution! SN2 inversion of configuration is also called Walden inversion, after Paul Walden (1863-1957), a pioneer in the study of substitution reactions. Inversion of configuration requires making and breaking of bonds, and therefore occurs only at the carbon bearing the leaving group. In the following example, inversion occurs at the carbon bearing the chlorine atom (the leaving group) but not the methyl group (not the leaving group). In this case inversion converts an R stereocenter into an S stereocenter, as well as trans into cis. Not inverted CH3 CH3 Na I Cl I Inverted (R)-2-Chloro-1-methyl- (S)-2-Iodo-1-methylcyclohexane (trans) cyclohexane (cis) In an SN2 reaction, why does backside attack occur? Repulsion of δ- charges on the nucleophile and leaving group is a simple explanation, but it fails to explain the cases where the nucleophile and leaving group have opposite charges in the transition state. An explanation that covers all SN2 reactions and is independent of the charges is this: the nucleophile attacks the σ* (sigma antibonding) orbital associated with the carbon-leaving group sigma bond. (Review bonding and antibonding orbitals from section 2.xx if necessary. Why these particular orbitals are involved is beyond the scope of our introductory exploration of ionic substitutions.) The nucleophile approaches from the backside, an orientation that leads to the Ionic Substitution Reactions – Page 10 greatest overlap between its lone pair orbital and the σ* orbital. (Recall that greater overlap gives stronger bonds, more stability, and in the case of a transition state, lower energy of activation.) lone pair Bonding and antibonding orbitals (2.xx). σ bond σ* nuc R1 R1 R1 nuc LG C R3 Review R3 R R2 nuc LG C + C LG R3 R2 2 Figure 11.4: Simplified scheme showing orbital involvement in the SN2 reaction. Concept Focus Question 11.10 Provide the product, mechanism and transition state for each of these SN2 reactions. Pay carefully attention to the stereochemistry. Label the stereocenter(s) of each reactant and product as R or S. Br I (a) Cl O Br + (c) H O (excess) CH3 H Br (b) I LiSCH3 (d) D H NaOCH3 H (D = deuterium = 2H) Concept Focus Question 11.11 Provide the missing electrophile for each SN2 reaction. Pay careful attention to stereochemistry. I (a) ??? H I (c) O CH3 O CH3 O ??? O SPh (b) ??? PhS (excess) (d) C6H12IS H3C S CH3 SPh Concept Focus Question 11.12 Does inversion occur in an SN2 reaction even if the carbon bearing the leaving group is not a stereocenter? As an example, consider the SN2 reaction of iodomethane with cyanide ion. Ionic Substitution Reactions – Page 11 Concept Focus Question 11.13 (a) From the view point of the Cahn-Ingold-Prelog priority rules (section 6.xx), explain why inversion of configuration converts almost any R stereocenter into S and S into R. (b) Every SN2 reaction occurs with inversion of configuration but does every SN2 reaction convert an R stereocenter to S (or S to R)? Design an example SN2 reaction to illustrate your discussion. Concept Focus Question 11.14 In order to explore the effect of leaving group and nucleophile charge on the SN2 transition state the following reaction was performed. NaN3 H H CH3 S(CH3)2 CH3 N3 (a) Draw the best Lewis structure for azide ion (N3-). (b) Draw the transition states and products expected from both backside and front side attack. Pay careful attention to the stereochemistry. (The product has a stereocenter.) (c) Which enantiomer of the product was formed? Concept Focus Question 11.15 Students often have trouble seeing inversion of configuration when an SN2 reaction occurs at the carbon of a cyclohexane ring. Has inversion occurred in the reaction shown below? By adding, subtracting or otherwise changing just one atom of the electrophile, rewrite the reaction so that inversion is now clear and obvious. I C N CN 11.3 Factors Influencing the SN2 Reaction You probably noticed in the previous sections and Concept Focus Questions that SN2 reactions include a wide variety of nucleophiles, electrophiles and leaving groups. Can we predict how these factors might influence an SN2 reaction, to make it faster, slower or perhaps not even occur at all? (Not every SN2 reaction that we might write is viable.) In this section we explore some important factors (or variables) and learn how to predict their effects on SN2 reaction rate and products. We will look at the nucleophile, the leaving group, the affect of groups attached to the carbon bearing the leaving group and the reaction solvent. Ionic Substitution Reactions – Page 12 nuc + R3C LG solvent nuc CR3 + LG Figure 11.5: The fundamental features that influence the rate and product of every SN2 reaction are the nucleophile (nuc), the leaving group (LG), the attachments to the carbon that bears the leaving group (CR3) and the solvent. A. The Nucleophile Figure 11.5 suggests two ways in which the nucleophile influences an SN2 reaction: product and rate. How does the nucleophile influence the product of an SN2 reaction? The nucleophile provides the new functional group. For example, if the nucleophile is cyanide ion (-C≡N), then the product is a nitrile (RC≡N). (Review Concept Focus Question 11.3 for more examples.) How does the nucleophile influence the rate of an SN2 reaction? A useful approach to this issue starts with the question “what is the role of a nucleophile in an SN2 reaction, and how does its structure influence its ability to carry out this role?” The role of the nucleophile is simple: to share an electron pair with the electrophile, resulting in a new covalent bond. Higher electron density makes the nucleophile less stable (more electron repulsion) therefore more strongly driven to share some of that electron density. (Don’t you have a greater desire to find a new place to live when your dorm room or apartment becomes too crowded?) Therefore any structural feature that influences electron density will also influence nucleophilicity. These structural features might include resonance, atomic radius, electronegativity, inductive effects and formal charge. Perhaps you are saying to yourself “Hey, this sounds familiar!” as you recall our previous studies on the relationship between basicity and electron density (sections 9.xx-9.xx). It is a good parallel: a nucleophile and a base both provide an electron pair for the new bond. They differ in the context of the bond formation: a nucleophile shares its electron pair with an electrophile, whereas a base shares its electron pair with the hydrogen of an acid. In other words, in a reaction where X shares its electron pair with Y to form a new X-Y bond, if Y is a hydrogen atom we call X a base. If Y is a carbon atom we call X a nucleophile. If this basicity/nucleophilicity parallel is true, then do the same features that influence basicity also influence nucleophilicity? The SN2 rate data of Table 11.1 will be useful to guide us to a conclusion. Ionic Substitution Reactions – Page 13 We measure nucleophilicity by the relative rate of SN2 reactions with various nucleophiles, and the same electrophile, solvent, concentration and temperature. Table 11.1: Relative rate for the SN2 reaction of selected nucleophiles with CH3Br in ethanol. Nuc H3C Nucleophile Br S CH3CH2OH H3C Nuc + Br I CH3CH2O HO Relative rate 57,000,000 120,000 60,000 12,000 H-Nuc pKa 7.8 -10 16.5 15.7 Nucleophile Br (CH3CH2)3N Cl Relative rate 5,000 2,000 1,400 1,100 H-Nuc pKa -9 10 10 -7 H2O F ClO4 O O Nucleophile CH3 O Relative rate 900 1 0 0 H-Nuc pKa 4.8 15.7 3.2 -10 Despite our prediction, however, the relative rate data in Table 11.1 make it obvious that the relationship between nucleophilicity and pK a is not universal. The problem here is that nucleophilicity and pK a relate to similar, but not identical reactions. A nucleophile bonds to carbon atom, whereas a base bonds to a hydrogen atom. Are there any trends at all in this data? Can we make any useful predictions about structure and nucleophilicity? 1. Resonance Review Structural effects on basicity (9.xx-9.xx). Think Ahead Question 11.5 Table 11.1 reveals that acetate ion (CH3CO2-) is a weaker nucleophile and weaker base than ethoxide ion (CH3CH2O-). What structural difference makes acetate ion a poorer base and a poorer nucleophile than ethoxide ion? It may be useful to review how structure influences basicity from sections 9.xx-9.xx. Answer: As we learned earlier, the mechanistic role of a nucleophile and a base is similar: to provide an electron pair to form a new covalent bond. We also predicted that the same structural features that influence basicity might also influence nucleophilicity. Table 11.1 tells us this parallel does not cover all Ionic Substitution Reactions – Page 14 nucleophiles, but it seems to apply in the case of acetate ion versus ethoxide ion. What structural difference makes acetate ion a poorer base and a poorer nucleophile than ethoxide ion? One obvious difference is resonance. Acetate ion has significant resonance stabilization and delocalization of its electron density. Ethoxide ion does not have this resonance delocalization. Its resonance hybrid is identical to its only significant resonance contributor. O CH3 O CH3 O CH3 Acetate ion has two significant resonance contributors. H H H C C H H X O Review O δ- O O δ- Resonance hybrid no additional resonance contributors Ethoxide ion has a single significant resonance contributor. Because of its resonance delocalization, each oxygen atom of acetate ion has less electron density than the oxygen of ethoxide ion. (Each oxygen atom of acetate ion bears a δ - charge whereas the oxygen of ethoxide ion has a full –1 formal charge.) Lower electron density reduces the drive to share electron density, so acetate ion is a poorer (less aggressive) nucleophile than ethoxide ion. This is exactly the same reason that acetate ion is a poorer base than ethoxide ion, as we learned in section 9.xx. Here is an alternate and equally valid explanation. We know that resonance is a stabilizing feature. A molecule that has resonance stabilization “wants” to keep it. A molecule that does not have resonance stabilization “wants” to get it. Upon forming a new covalent bond with the carbon atom of an electrophile such as methyl iodide, acetate ion suffers a reduction in the number of significant resonance contributors. This resonance loss destabilizes the transition state, and slows the reaction. Ethoxide does not suffer this resonance loss. Therefore acetate ion is a poorer nucleophile than ethoxide ion. O O O CH3I CH3 O CH3 O Before reaction: acetate ion has two significant resonance contributors CH3 CH3 O X no additional significant resonance contributors After reaction: the ester product has just one significant resonance contributor. A study of many other resonance-stabilized nucleophiles leads to this general rule: Ionic Substitution Reactions – Page 15 Reminder: When making a comparison between two or more things, focus on differences instead of similarities. Relative significance of resonance contributors (3.xx). General Rule Compared to similar nucleophiles without resonance, most nucleophiles with resonance delocalization of the electron pair used to form the new covalent bond with the electrophile are less nucleophilic. There are some exceptions to watch out for. Caution! Resonance does not always reduce electron density. A molecule may have resonance that does not influence the electron density at the atom that attacks the electrophile. In some rare circumstances resonance can even increase the electron density. (See Concept Focus Question 11.18 for more on this.) Concept Focus Question 11.16 Select the weaker nucleophile in each pair. Offer a brief explanation in each case. O O (a) and (b) NH2 and NH2 O O (c) O and NH2 (d) and NH2 Concept Focus Question 11.17 By adding, subtracting or otherwise changing at most three atoms, redraw each structure so that it has resonance (or even more resonance) that makes it a poorer nucleophile. Draw the resonance contributor(s) that account for the reduced nucleophilicity. (a) O (b) (CH3)3CCH2NH2 (c) O Concept Focus Question 11.18 It was mentioned previously that resonance does not always decrease nucleophilicity. With this thought in mind, select the faster reaction in each pair, and briefly explain why it is faster. Ionic Substitution Reactions – Page 16 H3C NH2 (a) H2N NH2 H2N CH2O (b) NH2 I CH3I H2N NH2 CH2O CH2OCH2CH3 versus NH I CH3I versus NH2 CH3CH2Cl H3C NH H2N NH2 CH2OCH2CH3 CH3CH2Cl Concept Focus Question 11.19 Write two SN2 products for this reaction. Hint: think about resonance. O CH3I A nucleophile that can react at more than one site on its structure is called an ambident nucleophile. 2. Electronegativity In the previous section we learned that resonance influences nucleophilicity in the same way it influences basicity. Is this also true for electronegativity? Think Ahead Question 11.6 Based on what you know about electronegativity effects on basicity (section 9.xx), rank these ions in order of increasing nucleophilicity: F-, HO- and H2N-. Answer: We have already established that basicity and nucleophilicity both involve sharing of an electron pair. Therefore we predicted that any structural feature that increases or decreases basicity has a parallel effect on nucleophilicity. Back in section 9.xx we deduced that electronegativity also influences basicity. Of the three ions in question fluoride ion is the weakest base because fluorine is more electronegative (i.e., less willing to share electron density) than oxygen or nitrogen. Oxygen is more electronegative than nitrogen, so the order of basicity is F- < HO- < H2N-. Therefore the order of nucleophilicity is F- < HO- < H2N-. Experiments conducted under conditions where no other factors interfere verify this prediction, and further show that the effect is general. General Rule The strength of a nucleophile is influenced by the electronegativity of the atom(s) that share an electron pair to form a covalent bond with the electrophile: higher electronegativity causes lower nucleophilicity. Ionic Substitution Reactions – Page 17 Review Electronegativity effects on basicity (9.xx). Concept Focus Question 11.20 For each reaction pair, select the faster reaction, and write its product. (a) Chloride ion and methanethiolate ion (CH3S-) reacting with 2-iodopropane. (b) Water and methylamine reacting with ethyl chloride. (c) Triphenylphosphine (Ph3P) and diphenylsulfide (Ph2S) reacting with (R)-1bromo-1-phenylethane. (d) NH and O reacting with methyl iodide. 3. Atomic Radius In section 9.xx we learned that basicity is influenced by atomic radius: larger atomic radius results in lower electron density and reduced basicity. Does this effect apply to nucleophilicity as well? Think Ahead Question 11.7 Methyl iodide was reacted with halide ions in DMF (the solvent) at 25oC. The relative SN2 reaction rates (krel) were found to be: F- krel > 7.5 (fastest), Cl- krel = 6.3, Br- krel = 3.3 and I- krel = 1.0 (slowest). What is the relationship between atomic radius and reaction rate? Suggest an origin for this effect. Review Effect of atomic radius on basicity (9.xx). Answer: These ions are all in the same family (column) of the periodic table. Recall from general chemistry that atomic radius increases as you move down the family. Fluoride ion is the smallest and most nucleophilic, whereas iodide is the largest and least nucleophilic. This atomic radius effect on nucleophilicity is parallel to what we learned previously (section 9.xx) in relationship to basicity: smaller atomic radius results in more concentrated electron density, and greater basicity (or nucleophilicity). Atomic radius also changes somewhat as you move across the row (period) of the periodic table. However, atomic radius differences within a period are much less than atomic radius differences within a family. When comparing atoms in the same column of the periodic table, atomic radius has more influence on nucleophilicity (and basicity) than electronegativity. When comparing atoms in the same row of the periodic table, electronegativity has more influence on nucleophilicity (and basicity) than atomic radius. (A more precise analysis uses ionic radius instead of atomic radius, but the qualitative results are the same.) Ionic Substitution Reactions – Page 18 Increasing electronegativity Same period: electronegativity dominates C 2.5 F 4.0 P 2.5 S 2.5 Cl 3.0 Se 2.4 Br 2.8 Increasing atomic radius O 3.5 Same family: atomic radius dominates N 3.0 I 2.5 Figure 11.6: The “northeast” corner of the periodic table showing trends in atomic radius (size of circles; not to scale) and electronegativity (number in the circle). A more rigorous analysis reveals that the origin of the effect is subtler and more complex than just electron density. (For example, Table 11.1 reveals that when the solvent is ethanol, iodide ion is a much more aggressive nucleophile than fluoride ion.) The atomic radius effect is also strongly influenced by how the solvent interacts with the reactants and transition state. Therefore we must reserve making a General Rule about atomic radius effects until we learn how polarizability (section 11.3C) and solvent (section 11.3E) influence an SN2 reaction. Caution! The atomic radius effect is based on the atomic radius of the atom that is sharing an electron pair with the electrophile, and not on the size of the entire nucleophile. Concept Focus Question 11.21 Select the strongest nucleophile in each set. Write its SN2 reaction with ethyl iodide. (a) CH3O- and CH3S(c) F- and HS(b) (CH3)3N and (CH3)3P (d) CH3O- and (CH3)3CO- Ionic Substitution Reactions – Page 19 4. Inductive Effects So far in this section we have seen that there is a good (although imperfect) parallel between the structural features that influence basicity and nucleophilicity. We have focused on the atom of the nucleophile that is sharing electron density with the electrophile. What happens if we make changes elsewhere in the nucleophile, away from the “business end”? Think Ahead Question 11.8 The data in Table 11.1 suggests that compared to other nucleophiles, acetate ion (CH3CO2-) is rather sluggish. Is trifluoroacetate ion (CF3CO2-) a stronger or weaker nucleophile than acetate ion? Answer: Acetate ion is a sluggish nucleophile because its electron density is delocalized by resonance, and because of the high electronegativity of oxygen. Trifluoroacetate ion has the same influences because it also a carboxylate ion (i.e., it has the same functional group). Review Inductive effect influence on basicity (9.xx). An obvious difference between the nucleophiles in question is the presence of a methyl group in CH3CO2- versus a trifluoromethyl group in CF3CO2-. How might the presence of fluorine influence nucleophilicity? When we think of fluorine one thing that comes to mind is its high electronegativity. Of all the atoms in the periodic table, fluorine has the greatest greed for electron density, and will take it from its neighboring atoms. (Hydrogen’s low electronegativity gives it no cause to steal or donate electron density to other parts of the molecule.) This theft of electron density from elsewhere in the molecule is called an inductive effect, and we have encountered it before when we studied basicity (section 9.xx). An electron-withdrawing inductive effect reduces basicity, and extending our parallel between basicity and nucleophilicity, we predict it also decreases nucleophilicity. Therefore CF3CO2- is a poorer nucleophile than CH3CO2-. Studies of many nucleophiles show that the inductive effect is general, so we can formulate a General Rule. General Rule Nucleophilicity is decreased by electron-withdrawing inductive effects, and increased by electron-donating inductive effects. Caution! Most, but not all, inductive effects that we will encounter are electron withdrawing in nature. A few are electron donating. Therefore do not assume that inductive effects always decrease nucleophilicity. Ionic Substitution Reactions – Page 20 Concept Focus Question 11.22 Select the weakest nucleophile in each set. (a) CF3CH2O- versus CH3CH2O- (c) FCH2CO2- versus FCH2CH2CO2(b) BrCH2CO2- versus Br3CCO2- (d) Cl2CHCO2- versus FCH2CO2Concept Focus Question 11.23 Select the major product of this reaction. O O F CH3I or F F OCH3 O CH3O O F F F Concept Focus Question 11.24 The data in Table 11.1 reveals that hydroxide ion is a substantially better nucleophile than perchlorate ion (ClO4-). What structural feature(s) account for this difference? Concept Focus Question 11.25 Measurement of SN2 reaction rates for some thiolate ions gives the following order of nucleophilicity: S < CH2S < CH2S What does this suggest about the inductive effect of a benzene ring? 5. Formal Charge In our studies of organic molecules as acids and bases we deduced that bases with formal negative charges are stronger than uncharged bases (section 9.xx). Does formal negative charge also enhance nucleophilicity? Think Ahead Question 11.9 The data in Table 11.1 reveals that hydroxide ion is a substantially better nucleophile than water. What structural feature(s) account for this difference? Review Formal charge effect on basicity (9.xx). Answer: Water and hydroxide ion do not have resonance to delocalize their electron density. In both cases an oxygen atom provides the electron pair that becomes the new covalent bond, so there is no difference in the atomic radius or electronegativity effects. Neither oxygen atom bears electron-withdrawing or electron-donating groups, so the nucleophilicity difference cannot be due to inductive effects. All that remains is the formal charge. How does formal charge influence electron density, our root cause of basicity and nucleophilicity? Back in section 9.xx we learned that a base with a formal negative charge is stronger than a neutral base, because a negative formal charge indicates the atom has one more electron than the corresponding uncharged atom of the same element. Keeping with our Ionic Substitution Reactions – Page 21 Review Meaning of formal charge (general chemistry and 1.xx). parallel between basicity and nucleophilicity, we therefore predict that a nucleophile with a formal negative charge is more nucleophilic than a nucleophile that is neutral. This explains why HO- is a stronger nucleophile (and stronger base) than H2O. General Rule Everything being equal, a nucleophile with a formal negative charge is more nucleophilic than a neutral (uncharged) nucleophile. If we write SN2 reactions for these nucleophiles with (for example) methyl iodide, we can draw a useful corollary of this formal charge effect. HO + H3C I HO CH3 + I Oxygen formal charge –1 → 0 Faster reaction H2O + H3C I H2O CH3 + I Oxygen formal charge 0 → +1 Slower reaction In these reactions, the oxygen atom of hydroxide ion (the better nucleophile) undergoes a formal charge change of –1 to 0 upon SN2 reaction. In the reaction of water (the weaker nucleophile) the formal charge of oxygen changes from neutral to +1 (it becomes an oxonium ion). Recall (section 1.xx) that atoms prefer to remain neutral. Therefore a formal charge change of negative to neutral enhances nucleophilicity whereas a formal charge change from neutral to positive decreases nucleophilicity. Concept Focus Question 11.26 For each pair of molecules, draw the conjugate base (review section 9.xx if necessary) and select the strongest nucleophile in each pair of conjugate bases. (a) NH3 versus +NH4 (b) Water versus methyl ether (c) H2S versus HS(d) Oxalic acid (HO2C-CO2H) versus hydronium ion (H3O+) 6. Relative Influence of Factors You may have noticed that in several of the Concept Focus Questions leading up to this point, different solutions were possible depending upon what assumptions you made about the relative influence or priority of the various factors that control nucleophilicity. It would be useful, therefore, to be able to rank the relative influence of these factors to help us understand and predict those cases where the factors are not synergistic. To assemble this ranking, we compare reaction rates for nucleophiles that have two factors in conflict, such as atomic radius and electronegativity, but all other factors equal. Think Ahead Question 11.10 gives you a taste of how this might be done. Ionic Substitution Reactions – Page 22 Think Ahead Question 11.10 Using the relative rate data given in Table 11.1, decide which of the structural factors that influence nucleophilicity is dominant: (a) CH3CH2O- versus Cl-, and (b) CH3CO2- versus H2O. Answer: Let’s start by laying out our tool kit (so to speak), with a list of the structural features that we have deduced so far: • • • • • Resonance Electronegativity Atomic radius Inductive effect Formal charge In continuing our parallel between nucleophilicity and basicity, note that this is identical to the list of features that influence basicity (section 9.xx). Now let’s apply these to the nucleophile pairs in question. CH3CH2O- is more nucleophilic than Cl-. The factors at odds in this pair are electronegativity and atomic radius. The fact that ethoxide ion is a stronger nucleophile than chloride ion suggests that the smaller atomic radius of oxygen carries more weight than the lower electronegativity of chlorine. Therefore in terms of relative influence we can say atomic radius > electronegativity. CH3CO2- is more nucleophilic than H2O. The factors at odds in this pair are resonance and formal charge. The fact that acetate ion is a stronger nucleophile than water suggests that the formal negative charge of acetate has a greater effect on its nucleophilicity than its resonance. Therefore in terms of relative influence we can say formal charge > resonance. Considering many nucleophile pairs like this reveals a general trend of influences, which we use as the basis for a General Rule. General Rule The relative importance of structural features that influence nucleophilicity is: Resonance > atomic radius > electronegativity > inductive effects The influence of formal charge varies greatly, sometimes carrying more weight than resonance and in other cases, less. Therefore we cannot rank it with any certainty. There are exceptions to this ranking, but it makes accurate predictions often enough to be useful. (We will explore some of these exceptions later in this chapter.) Our theory of nucleophilicity would need to be more complex to cover every case and every nuance. A simple, easily understood theory that gets it right most of the time is very useful. (A good professional baseball player may only Ionic Substitution Reactions – Page 23 get on base 35% of the time, and look how much he gets paid! Imagine how much more he would get if he got it right 80% of the time.) Since we are discussing nucleophilicity factors as a group, this is a good time to mention a pitfall experienced by some students. Caution! When applying the nucleophilicity factors, all but inductive effects apply to the atom(s) that form the new covalent bond with the electrophile. (You might think of these atoms as the “business end” or “warhead” functional group of the nucleophile.) If the portion of the molecule under question is not part of the functional group that becomes bonded to the electrophile, then it can only influence nucleophilicity through an inductive effect. Resonance Atomic radius operate Electronegativity here Formal charge Inductive effect operates here X Y R3C LG SN2 X Y CR3 + LG Concept Focus Question 11.27 Select the strongest nucleophile in each pair. Briefly explain your reasoning. (a) CF3CH2O- versus CH3CO2(b) The conjugate bases of HOCl and HOBr (c) Aniline and cyclohexanol Concept Focus Question 11.28 By adding, subtracting or otherwise changing no more than three atoms, modify each of the strongest nucleophiles from Concept Focus Question 11.27 so that it is even stronger. B. The Leaving Group Now we turn our attention to the leaving group, the portion of the electrophile that breaks away along with the electron pair that used to be the carbon-leaving group bond. What constitutes a good leaving group? The role of the leaving group appears to be exactly opposite that of the nucleophile. The leaving group accepts an electron pair as the carbon-leaving group bond breaks whereas a nucleophile supplies an electron pair as the carbonnucleophile bond forms. The more effectively the leaving group can accommodate its new electron pair the more readily it will leave. (In more rigorous terms, a more stable leaving group reduces the energy of activation and makes the reaction faster.) Whether we are concerned about nucleophiles or Ionic Substitution Reactions – Page 24 leaving groups, it appears that ability to stabilize electron density is the central issue. Do the same features that influence electron density for a nucleophile also operate for a leaving group? Think Ahead Question 11.11 Consider these relative rates of the following reaction pairs. Does this data support the idea that the factors that control nucleophilicity also operate for leaving groups? methanesulfonate O O (a) H3C O S LiSCH3 CH3 H3C SCH3 + O H3C H3C H3 C I Cl CH3 Faster O O (b) S OH LiSCH3 LiSCH3 H3C LiSCH3 H3 C H3C SCH3 + OH SCH3 + I Faster SCH3 + Cl Slower No reaction (very slow) Answer: The role of the leaving group is to depart along with an electron pair. The more effectively the leaving group can accommodate or stabilize the electron pair it gains, the more readily it leaves. Methanesulfonate ion is a better leaving group than hydroxide ion: The electron pair ends up on an oxygen atom. Oxygen is highly electronegative so this assists the leaving group to accommodate the new electron pair. Each leaving group departs with a negative formal charge. Recall that atoms prefer to have zero formal charge, so this factor slows the leaving group’s departure. Both leaving groups are influenced equally by these factors, so they are not the factors that account for the difference in leaving group ability. So what structural difference operates here? When it leaves, methanesulfonate ion gains significant resonance to delocalize the pair of electrons it gains upon departure (Figure 11.7), whereas hydroxide ion does not. As we have seen many times before, resonance is a stabilizing feature. Methanesulfonate can accommodate and disperse the new electron pair through resonance more effectively than hydroxide ion, so methanesulfonate ion is a better leaving group. (This is very much like sulfuric acid, which is very strongly acidic due to the resonance it gains upon deprotonation. Review section 9.xx.) Ionic Substitution Reactions – Page 25 O Methanesulfonate ion: O O S CH3 O O Hydroxide ion: S O CH3 O O H X O S CH3 O No additional resonance contributors Figure 11.7: Methanesulfonate has significant resonance stabilization but hydroxide ion does not. Iodide ion is a better leaving group than chloride ion: Iodide ion has a larger radius than chloride ion, and therefore more space to accommodate the new electron pair (less repulsion). A broader study of leaving groups reveals that same the factors that influence nucleophilicity (and basicity) also apply to leaving group ability, but in a reverse sense. A factor that enhances nucleophilicity tends to inhibit leaving group ability. However, this reversal is not perfect, because we are ignoring other factors such as how the solvent influences the nucleophile and leaving group. On the other hand, we can categorize leaving groups into four approximate categories: good, moderate, rare and never (Table 11.2). The characterization is based on empirical data, but can be readily understood by applying the five structural features we have used so many times before. (It is fairly easy to assemble this list for leaving groups because the collection of common leaving groups is small. It would be nice to have such a list for nucleophiles, but there are too many common nucleophiles to make the list useful.) Ionic Substitution Reactions – Page 26 Table 11.2: Scale of leaving groups O O R O S O CH3 S Sulfonate O O R Decreasing leaving group strength CH3 I Iodide I R Br Br R Cl Cl Bromide Chloride H R Excellent leaving groups Water H2O O H About equal Moderate leaving groups CH3 R Alcohol CH3OH O H R F Fluoride F O O Carboxylate* R O O CH3 R N(CH3)3 N(CH3)3 OCH3 OCH3 R R R NH2 NH2 R R OH OH H CH3 H CH3 CH3 Rarely leave Amine Alkoxide Hydroxide About equal Leave only under special circumstances (25.xx and 28.xx) Nitranion Hydride Only in Chichibabin reaction (24.xx) Carbanion Never leaves unless stabilized (26.xx and 28.xx) *Carboxylates often suffer nucleophilic attack at the carbonyl carbon more readily than they function as leaving groups. For more detail see section 27.xx. Ionic Substitution Reactions – Page 27 A s u l f o n a t e has the general structure ROSO2R’ or RSO3-. Departure of a leaving group is a common mechanistic step, and we will encounter it in many other reactions besides SN2. The leaving group propensities of Table 11.2 apply to these other reactions as well. Caution! Despite the general relationships between basicity, nucleophilicity and leaving group ability, not all good nucleophiles are bad leaving groups. For example, iodide ion is a good nucleophile as well as an excellent leaving group. (These exceptions exist because the basicity/nucleophilicity/leaving group relationship is not perfect.) Concept Focus Question 11.29 For each pair of molecules, select the best leaving group and offer a brief explanation for your choice: (a) Water versus hydroxide ion; (b) fluoride ion versus bromide ion; and (c) hydroxide ion versus acetate ion. Concept Focus Question 11.30 Phosphate ions such as diphosphate are leaving groups in biochemical reactions. Would you categorize diphosphate as a good, moderate or poor leaving group? O R O P O O P O O O O O P O O P O O O Diphosphate Concept Focus Question 11.31 Sulfonate leaving groups with different structures have been developed for various purposes. Rank the following sulfonate ions in terms of leaving group ability. Briefly explain your reasoning. (You can learn more about sulfonates and other strategies to improve an OH or OR leaving group in section 20.xx.) O CH3 S O O CF3 O S O - Methanesulfonate (mesylate; OMs) Trifluoromethanesulfonate (triflate; -OTf) O CH3 O S O O Br O S O O - p-Toluenesulfonate (tosylate; OTs) p-Bromobenzenesulfonate (brosylate; -OBs) Concept Focus Question 11.32 Explain why there is little if any difference in the leaving group ability of water and of an alcohol such as methanol. Ionic Substitution Reactions – Page 28 Concept Focus Question 11.33 Table 11.2 reveals carbanions are usually not leaving groups. However, a triiodomethyl carbanion or an enolate can leave in certain reactions. What do these leaving groups have that make them an exception to the carbanion leaving group rule? O I C I I Triiodomethyl carbanion: leaving group in the iodoform reaction (section 26.xx) Enolate: leaving group in the retroaldol reaction (section 28.xx) C. A Few Words About Polarizability Now that we have some experience with nucleophiles and leaving groups, it seems appropriate to briefly explore a structural feature that is the root of some of the exceptions that have come up. Polarizability is simply the ease with which an electron cloud can be distorted. (A rigorous definition and understanding of its origin is beyond our introductory explorations in organic chemistry, but can be found in most any advanced organic chemistry textbook.) An atom or molecule is called soft if its electron cloud is easily distorted, and hard if electron cloud distortion is more difficult. Large atomic radii and little or no charge characterize soft species. Hard species feature smaller atoms and large charges. The sulfur atom of hydrogen sulfide is soft because sulfur is a third row element (large atomic radius) and it has no formal charge. Fluoride ion is hard because it is small (the smallest second row element) and has a formal charge of –1. How does this apply to SN2 reactions? An SN2 reaction involves two bond change events that require electron clouds to change: formation of a nucleophile-carbon bond and rupture of a carbon-leaving group bond. (Any curved arrow represents an electron cloud change of some sort.) When the electrons clouds involved are easily distorted, the reaction occurs more readily (more stable transition state and lower energy of activation), as shown in Figure 11.8. Therefore we predict that SN2 reaction rate might be enhanced when the atoms that are forming or breaking bonds to the carbon bearing the leaving group are soft. R nuc C R LG R Polarization of these electron clouds stabilizes the SN2 transition state Figure 11.8: Polarizability effects on the SN2 transition state. Ionic Substitution Reactions – Page 29 Polarizability influences many other molecular interactions. For example, harder atoms make for stronger bases (section 9.xx) and stronger hydrogen bonds (section 7.xx). This prediction has been verified in the laboratory, and it helps explain a few apparent exceptions we have encountered. For example, iodide is a good nucleophile as well as an exceptional leaving group because it is very soft. Its electron cloud eagerly reaches out to carbon when iodide is the nucleophile, and readily withdraws away from carbon when it is the leaving group. Concept Focus Question 11.34 Categorize these nucleophiles as hard or soft: (a) hydride ion, (b) hydroxide ion, (c) triphenylphosphine (Ph3P) and (d) bromide ion. Concept Focus Question 11.35 By adding, subtracting or otherwise changing exactly one atom in each case, rewrite each SN2 reaction so that it is faster because it has a softer leaving group. LiOH (a) CH3F (b) CH3CH2OH2 (c) CH3OH KF CH3CH2F NaH CH2N(CH3)3 CH3 D. Steric Effects So far in this section we have explored the nucleophile and the leaving group, two of the four SN2 reaction variables that we previously identified. Now we ask what happens when we change the groups attached to the carbon bearing the leaving group. Think Ahead Question 11.12 In order to ascertain the effect of changing groups at the carbon bearing the leaving group, a series of alkyl bromides RBr was reacted with chloride ion in DMF (a solvent) at 25oC. Use this rate data to formulate a statement concerning the effect of increasing the number of methyl groups at the carbon bearing the leaving group. Explain the origin of the trend. Hint: Use your molecular models, with a small ball for the nucleophile. R Br + Cl R DMF, 25oC Cl + Br CH3 CH3 CH3 Br CH3CH2 Br H C RBr CH3 Relative rate 2000 60 1 Br CH3 C Br CH3 ~0 * *The relative SN2 rate for tert-butyl bromide is so slow that an alternate ionic substitution mechanism takes over (section 11.6). Ionic Substitution Reactions – Page 30 Answer: The effect of increasing the number of methyl groups at the carbon bearing the bromine atom is straightforward: more methyl groups cause a slower reaction rate. We will see many other situations in which the number of carboncarbon bonds influences a reaction, so it is useful to have a naming system that describes the number of these bonds. In this system, a carbon is labeled based on the number of carbons immediately attached to it. A carbon that is not bonded to any other carbons is called a methyl carbon. One attachment makes it a primary carbon (also called 1o carbon), two is a secondary carbon (2o), three is a tertiary carbon (3o), and four is a quaternary carbon (4o). Category Methyl Skeleton Primary 1o C C C Secondary 2o Tertiary 3o Quaternary 4o C C C C C C C C C C C C This carbon categorization is based on carbon-carbon bonds only. For example, consider the carbons of CH3CH2Br. The CH3 carbon is attached to one other carbon (the CH2) so it is a primary carbon. The CH2 carbon is also primary because it is attached to just one other carbon (the CH3). The bromine atom is not included. Concept Focus Question 11.36 Label each carbon as methyl, primary, secondary, tertiary or quaternary. (CH3)2N (a) CH3O H H CH3 CH3 C C C H H CH3 CH3 C CH3 (b) With our new vocabulary in hand, we can rephrase Think Ahead Question 11.12: Why is an SN2 reaction at a primary carbon slower than the same reaction at a methyl carbon? How does the extra methyl group cause the reaction to be slower? Since we are talking about an effect on reaction rate, the answer lies in the energy of activation, the energy difference between the reactants and transition state. This is where your models come in. Let’s start with the methyl bromide case. Use the model and ball to visualize what happens as the nucleophile approaches the carbon bearing the methyl group. As the nucleophile approaches the backside of the carbon-bromine bond (recall backside attack from section 11.2B), van der Waals repulsion (a nonbonded interaction; section 5.xx) begins to build between the nucleophile and the hydrogens. The hydrogens respond by moving away, eventually becoming approximately coplanar with the carbon atom in the transition state (section Ionic Substitution Reactions – Page 31 Review Van der Waals (nonbonded) interactions (5.xx). 11.2B). The repulsions still occur in the transition state, and are finally minimized when the product is formed. In the transition state, the hydrogens have moved closer to the leaving group, causing hydrogen-bromine interactions. All of these van der Waals interactions raise the energy as the molecule moves into the transition state. This process is illustrated in Figure 11.9. Cl- + H3C-Br Review Space-filling models (5.xx). Transition state Cl-CH3 + Br- Figure 11.9: Space-filling models for the CH3Br + Cl- SN2 reaction showing van der Waals (nonbonded) interactions. As chloride ion approaches the backside of the carbon-bromine bond, the hydrogen atoms shift in response to nonbonding interactions with the incoming nucleophile. Color scheme: orange = Cl, red = Br, black = C and white = H. The magnitude of nonbonding interactions is influenced by the size of groups involved. Larger groups tend to bring their electron clouds closer together, resulting in stronger repulsion (section 5.xx). In these SN2 transition states, as we replace hydrogen atoms (small) with methyl groups (larger), repulsions become more severe, resulting in increased transition state instability and slower reactions. Thus in the SN2 reaction with chloride ion, methyl bromide undergoes substitution most readily and tert-butyl bromide is very slow (almost inert). CH3CH2Br + Cl- (CH3)2CHBr + Cl- (CH3)3CBr + Cl- Figure 11.10: Space-filling models for R3CBr + Cl- SN2 transition states. Nonbonding interactions become more severe with an increasing number of methyl groups. Color scheme: orange = Cl, red = Br, black = C and white = H. Review Steric effect (5.xx). This is an example of a steric effect (section 5.xx). Steric hindrance is a steric effect that prevents atoms or groups from approaching each other. In this case chloride ion suffers steric hindrance in approaching the backside of the carbonbromine bond, especially in tert-butyl bromide, (CH3)3CBr. Steric hindrance is a common phenomenon in organic reactions. Steric hindrance explains the relative rate data of Think Ahead Question 11.12: methyl bromide reacts fastest because it has the least severe nonbonding interactions (lowest steric hindrance) in the transition state. The interactions Ionic Substitution Reactions – Page 32 increase (and reaction rates decrease) with higher degree of substitution at the carbon bearing the leaving group. Studies of a wide range of SN2 reactions have shown this is a general trend, so we can formulate a General Rule concerning the role of steric hindrance in an SN2 reaction. General Rule In an SN2 reaction, increasing the degree of substitution at the carbon bearing the leaving group slows the reaction. H H C H LG > R H C R LG > H Methyl R C R LG > H Primary Secondary R C LG R Tertiary Decreasing rate of SN2 reaction A final note before we turn to some Concept Focus Questions: an SN2 reaction occurs only at an sp3 carbon. Substitution reactions at sp2 carbons occur by other mechanisms. (For substitution at aromatic carbons see Chapter 24. For substitution at carbonyl carbons, see Chapters 25-27.) Concept Focus Question 11.37 (a) Categorize each carbon bearing a leaving group as methyl, primary, secondary or tertiary: ethyl iodide, methyl fluoride, cyclohexyl chloride and tert-butyl triflate, (CH3)3COSO2CF3. (b) Select the molecules in part (a) that would have the fastest and slowest SN2 reactions. Concept Focus Question 11.38 Explain the following SN2 relative rate data. Hint: build molecular models, and consider the reaction from the nucleophile’s perspective. RBr krel CH3CH2Br CH3CH2CH2Br (CH3)2CHCH2Br (CH3)3CCH2Br Ethyl bromide Propyl bromide Isobutyl bromide Neopentyl bromide 1.0 0.41 1.2 x 10-3 1.2 x 10-5 Concept Focus Question 11.39 Why don’t we bother to discuss the rate of an SN2 reaction at a quaternary carbon? Ionic Substitution Reactions – Page 33 Concept Focus Question 11.40 Explain why an SN2 reaction in which the nucleophile is tert-butoxide ion, (CH3)3CO-, is slower than a similar reaction in which methoxide ion, CH3O-, is the nucleophile. E. Solvent Effects So far in this chapter we have explored three reaction variables that influence the rate and outcome of an SN2 reaction: the nucleophile (section 11.3A), the leaving group (section 11.3B) and steric effects (section 11.3D). Now we turn our attention to the role of the solvent. You may be familiar with the term “solvent” as a liquid used to dissolve something else, such as the hot water used to dissolve freeze-dried coffee powder (and perhaps sugar) to make a cup of coffee. The role of solvent in a chemical reaction is similar, but also more complex. If we want to conduct a reaction between two substances that are solids, they will not react if we just mix the solids together. In the solid state the reactants cannot mingle and move to achieve the appropriate orientation for the reaction to occur. In the solution phase, the reactants are free to move about as needed. Solvents may also serve many other roles, such as rapid dispersion of heat, precipitation of a product in order to shift the reaction equilibrium towards the products, etc. The solvent may also be a reactant and thus become part of the product. Such reactions are called solvolysis reactions. Most of these uses are practical considerations that are important in lab work but beyond the scope of our introductory explorations. We can gain useful insight, however, by considering the effect of solvent on SN2 reactions. What we learn here can be applied to other organic reactions as well. Let us start the exploration with a list of common laboratory solvents, Table 11.3. The list includes proticity (ability to donate a hydrogen for hydrogen bonding) and dielectric constant (ε; ability to insulate opposite charges), two important properties that will concern us later in this section. Ionic Substitution Reactions – Page 34 Table 11.3: Structures and Properties of Common Solvents Polar solvents (ε > 20) Name Water Structure Proticity Dielectric constant O protic 80 aprotic 49 aprotic 38 aprotic 37 protic 33 protic 25 aprotic 21 aprotic 9.1 aprotic 7.6 protic 6.2 H Dimethylsulfoxide (DMSO) Acetonitrile H O S CH3 CH3 CH3 N,N-Dimethylformamide (DMF) C N O H N(CH3)2 Methanol CH3 Ethanol CH3CH2 OH OH O Acetone CH3 CH3 Nonpolar solvents (ε < 20) Dichloromethane ClCH2Cl Tetrahydrofuran (THF) Acetic acid (HOAc) O O CH3 OH Ethyl ether CH3CH2OCH2CH3 aprotic 4.3 Hexane CH3(CH2)4CH3 aprotic 1.9 So which solvent should we pick for an SN2 reaction? One criterion is reaction rate: choose a solvent that makes the reaction fastest. How does the solvent influence the reaction rate? The Arrhenius equation (section 8.xx) tells us that reaction rate is an exponential function of the energy difference (Ea) between the reactants and transition state. The larger this energy difference is the slower the reaction proceeds. Any factor in may alter this energy difference, and thus influence the reaction rate. For example, if the energy gap is smaller in solvent A than in solvent B, then the reaction is faster in solvent A. Compared to solvent A, Ionic Substitution Reactions – Page 35 Review Arrhenius equation (8.xx). solvent B provides good stabilization for reactants but does little to stabilize the transition state, so solvent B causes a larger energy gap and a slower reaction (Figure 11.11). Therefore we need to understand how a solvent influences the stability of the reactants and transition state. [TS] [TS] Ea (solvent B) Energy Energy Ea (solvent A) reactants reactants products products Reaction coordinate Energy profile in solvent A Reaction coordinate Energy profile in solvent B Figure 11.11: Energy of activation is dependent upon solvent. Compared to solvent A, solvent B offers significant stabilization for the reactants but little for the transition state. Energy of activation (solvent A) < energy of activation (solvent B), so rate of reaction A > rate of reaction B. Interactions between molecules in solution are very complex, but we can gain a very good understanding – sufficient for SN2 as well as most other organic reactions – by focusing on just two solvent properties: proticity and dielectric constant. Review Hydrogen bonding (7.xx). Review Bond polarity (1.xx). Proticity is the solvent’s ability to donate a hydrogen atom for a hydrogen bond. As we learned in section 7.xx, a hydrogen bond donor has a hydrogen atom with a large δ+ charge because it is bonded to a highly electronegative atom (F-H, N-H or O-H bonds). Looking at Table 11.3 we see that only O-H bonds are important among the common solvents. A solvent that has an O-H bond, and therefore can donate a hydrogen bond, is called protic. Methanol (CH3OH) and ethanol (CH3CH2OH) are important protic solvents. A solvent that cannot donate a hydrogen bond is called aprotic (“not protic”). DMF and DMSO are important aprotic solvents. (Remember: protic and aprotic refer to the solvent’s ability to donate, not to accept, a hydrogen bond.) Dielectric constant (ε) is the measure of a substance’s ability to insulate opposite charges from each other. When ε > 20 we say the solvent is polar, and when ε < 20 it is nonpolar. (The ε = 20 dividing line is a matter of convenience. There is not a sudden change in solvent behavior when ε crosses this line.) The number and intensity of δ+ and δ- charges on the solvent molecules determine the solvent polarity. Dielectric constant is higher when a solvent has many, strongly polar bonds because regions of high δ + and δ- interact more strongly with δ + and δ charges on other molecules. Water has a high dielectric constant (ε = 80) because it has two very polar bonds and no nonpolar bonds. Methanol (ε = 33) is more polar than ethanol (ε = 25) because both have two polar bonds (C-O and C-H) but methanol has less nonpolar C-C and C-H bonds than ethanol. Ionic Substitution Reactions – Page 36 Concept Focus Question 11.41 Assign these dielectric constants (35, 20, 13.9, 13.3 and 10.3) to these molecules: 1-propanol, 1,3-propanediol, 1-pentanol, 1-hexanol and 1-octanol. Concept Focus Question 11.42 In light of its many polar bonds, acetic acid has a surprisingly low polarity (ε = 6.2). Suggest a reason for this. Hint: think about hydrogen bonding in pure acetic acid. How do solvent molecules interact with reactants and transition states? Think Ahead Question 11.13 Considering the fact that opposite charges attract and like charges repel, draw a picture that shows how molecules of methanol surround and interact with a chlorine ion, a molecule of methyl bromide, and the Cl-/CH3Br SN2 transition state. (Review Chapter 7 if your understanding of noncovalent molecular forces has faded.) Answer: Methanol molecules interact with Cl- and CH3Br through hydrogen bonding and dipole-dipole forces. (Van der Waals forces also operate, but these are much weaker so we can ignore them for our simplistic analysis.) The molecules orient themselves so as to place opposite charges adjacent and like charges pointing away from each other. Chloride ions (anions) are attracted to the δ+ H of methanol’s O-H bond, forming H||||||Cl hydrogen bonds (Figure 11.12). The association of a molecule with one or more molecules of solvent is called solvation, and the group of solvent molecules closely associated with the molecule is called the solvent shell. Solvation of Cl- by methanol is a stabilizing interaction because the H δ+ helps counteract the formal –1 charge of chloride ion. Figure 11.12: Interaction of methanol (solvent) molecules with a chloride ion. The left picture shows the atomic positions (orange = Cl, red = O, white = H and black = C.) The right picture shows the interactions of the charges (red = more negative and blue = more positive). One chloride ion is surrounded by more methanol molecules than are shown. The structure is not as highly organized as shown. Ionic Substitution Reactions – Page 37 Review Noncovalent molecular forces (Chapter 7). Methyl bromide does not have any strong charges, so its interaction with methanol is weak and less strongly organized. (Methanol molecules are attracted to each other more strongly than they are attracted to methyl bromide.) Compared to methyl bromide in the absence of any solvent, methyl bromide receives little stabilization through solvation by methanol. The transition state has δ - charges on Cl and Br. All other atoms are neutral. These δ- charges attract hydrogen bonds from methanol, but the attractive forces are not very strong. Therefore the transition state receives less stabilization by solvation than does Cl-, but more solvent stabilization than does CH3Br. The combination of these solvation interactions provides more stabilization to the reactants than it does to the transition state. Compared to the absence of solvent, this causes an increase Ea, and a slower reaction. Our goal is to select a solvent to make the reaction faster, so we need one that decreases Ea: a solvent that stabilizes the weakly charged transition state more than the highly charged reactant. A nonpolar solvent such as hexane meets this requirement. However, hexane (and most other nonpolar solvents as well) is not a practical choice because nonpolar solvents do not dissolve ionic reactants such as Cl-. Only polar solvents like DMF or CH3OH do this effectively. Therefore we usually have no choice but to use polar solvents for SN2 reactions. (Nonpolar solvents can be used with uncharged reactants.) In the exploration of solvent polarity and reaction rate, we used a reaction with a negatively charged nucleophile (Cl-) and a neutral electrophile (CH3Br). Most SN2 reactions involve a negatively charged nucleophile and neutral electrophile, but other charge combinations exist. In Concept Focus Question 11.43 we use the same ideas about solvent polarity and stabilization to predict solvent effects on these reactions as well. Concept Focus Question 11.43 Draw the transition state and products for each of the following SN2 reactions. Decide if the reaction is faster in DMF or THF. (a) CH3S + CH3I (c) NH3 + CH3I (b) NH3 + CH3S(CH3)2 (d) CH3S + CH3S(CH3)2 In these past few paragraphs we have explored the relationship of reaction rate and solvent polarity (as measured by dielectric constant). Now we turn our attention to the effect of proticity on reaction rate. Think Ahead Question 11.14 Consider this rate data, which shows the effect of CH3OH (protic solvent) versus DMF (aprotic solvent) on the SN2 reaction of methyl iodide with halide ion nucleophiles. Ionic Substitution Reactions – Page 38 Nuc: + H3C-I 25o C solvent DMF (aprotic; ε = 37) Nuc CH3 + I CH3OH (protic; ε = 33) Nucleophile k, M-1 s-1 log k k, M-1 s-1 log k FClBrI- >3 2.5 1.3 0.4 > 0.48 0.40 0.11 -0.40 5.0 x 10-8 3.0 x 10-6 8.0 x 10-5 3.4 x 10-3 -7.3 -5.5 -4.1 -2.5 Prepare a plot of log k for the various nucleophiles in both solvents. Use this plot as a guide to explain why all halides are stronger nucleophiles in DMF than in CH3OH. Answer: The completed graph (Figure 11.13) reveals several interesting trends, including the fact that all of the halide ions are better nucleophiles in DMF than in methanol. What difference in solvent properties of DMF and methanol account for this? Table 11.3 reveals DMF (ε = 37) is a bit more polar than CH3OH (ε = 33). This small difference in polarity does not see adequate to account for the huge difference in nucleophilicity. In what other ways are DMF and methanol different? 1 F- DMF CH3OH Cl0 BrI- -1 -2 log k I-3 -4 Br- -5 Cl-6 -7 F- Figure 11.13: Effect of solvent (DMF versus CH3OH) on the SN2 reaction rate of halide ion nucleophiles. Table 11.3 also reminds us that DMF is aprotic and CH3OH is protic. This difference encourages us to ask how does hydrogen bonding influence nucleophilicity? For an anion like fluoride ion, hydrogen bonding provides stabilization by dispersing some of the anion’s negative charge. An aprotic Ionic Substitution Reactions – Page 39 solvent does not provide this form of stabilization. A hydrogen bonded solvent shell of CH3OH surrounds F- (much like paparazzi surround a celebrity). In order to get close enough to form a bond with the electrophile, F- must shed some of this stabilizing solvent shell (Figure 11.14). This reduction in stabilization is felt in the transition state, causing an increase in the energy of activation and a slower reaction. (The celebrity must shed the clinging shell of paparazzi to enter the awards show.) The energy of activation cost for desolvation of F- is so great that F- is useless as a nucleophile in protic solvents. = CH3OH = F- = CH3I Figure 11.14: In order for F- and CH3I to reach the SN2 transition state, some CH3OH molecules (arrows) of the F- solvent shell must move out of the way. This desolvation reduces the stability of F-, raises the energy of activation and slows the reaction. Compare this with a celebrity surrounded by their paparazzi “solvation shell.” Any negatively charged nucleophile in a protic solvent will need to shed a portion of its solvent shell to achieve the transition state, so compared to an aprotic solvent, any negatively charged species is less nucleophilic in a protic solvent. Therefore an SN2 reaction will be faster in an aprotic solvent than in a protic solvent of comparable polarity. Compared to charged nucleophiles, a protic solvent does not significantly impact the nucleophilicity of nucleophiles without a formal charge because hydrogen bonds to charged nucleophiles are usually stronger than hydrogen bonds to neutral nucleophiles. (Review hydrogen bonding from section 7.xx.) Now we can formulate a General Rule concerning solvent choice for SN2 reactions. General Rule For most SN2 reactions, a polar aprotic solvent is preferred. Now we can explain another trend in solvent effects on nucleophilicity that is revealed by Figure 11.13. Ionic Substitution Reactions – Page 40 Think Ahead Question 11.15 Figure 11.13 reveals that fluoride ion loses much more nucleophilicity in a protic solvent than iodide ion does. Explain. Answer: We learned in the previous section that hydrogen bonding is the cause of reduced nucleophilicity in protic solvents. The data in Figure 11.13 tells us that fluoride ion loses much more nucleophilicity in CH3OH than iodide ion does. Is this because fluoride ion forms stronger hydrogen bonds than iodide ion? Hydrogen bonding is an electrostatic attraction of a δ+ H with one or more lone pairs on the acceptor atom. The force of this attraction is a function of the concentration of the charges: greater charge concentration causes stronger attraction. (You may recognize this as a consequence of Coulomb’s law, which you may have studied in an introductory physics course.) Fluoride ion (ionic radius 1.19 Å) is smaller than iodide ion (ionic radius 1.67 Å) but they both have the same –1 formal charge. Therefore the negative charge of fluoride ion is more concentrated (more charge per unit volume) than iodide ion, so fluoride ion forms stronger hydrogen bonds than iodide ion. Stronger hydrogen bonds make it harder for the nucleophile to desolvate as it approaches the transition state, making for slower SN2 reactions. Chloride and bromide ions fit this trend nicely. Concept Focus Question 11.44 Select the faster SN2 reaction in each pair. (a) Benzyl iodide (PhCH2I) plus LiCl in methanol versus DMF (b) Benzyl chloride in DMSO reacting with NaF versus NaBr (c) Benzyl chloride reacting with NaI in water versus acetone Concept Focus Question 11.45 Other nucleophiles besides halide ions have differing nucleophilicity due to their relative atomic radii. Find a pair of nucleophiles in Table 11.1 that also display this effect. Concept Focus Question 11.46 Polarizability also influences bond strength. In general, the bond between two hard atoms is stronger than a bond between a hard atom and a soft atom. Based on polarizability (sections 7.xx and 11.3C) explain why fluoride ion forms stronger hydrogen bonds than iodide ion. Concept Focus Question 11.47 (a) DMSO has a surprisingly high dielectric constant (ε = 49) given that it has just one polar bond (S=O) among eight nonpolar bonds (H-C and C-S). Suggest reason why DMSO is very effective at separating anions from cations. Hint: build a model and see how it interacts with a ball that represents a cation or an anion. (b) Based on what you have concluded about the dielectric constant of DMSO, do you expect hexamethylphosphoramide to be polar or nonpolar? (HMPA was used as an ionic substitution reaction solvent at one time but it has dropped out of use because it is carcinogenic to laboratory animals.) Ionic Substitution Reactions – Page 41 O (CH3)2N P N(CH3)2 (CH3)2N Hexamethylphosphoramide (HMPA) (c) Dimethoxyethane (DME; CH3OCH2CH2OCH3) and ethyl ether (CH3CH2OCH2CH3) are both ethers, but SN2 reactions of lithium iodide are millions of times faster in DME than in ethyl ether. Suggest a reason. 11.4 Reversibility of the SN2 Reaction At this point in your studies, you have encountered dozens of SN2 reactions (or maybe more if your are a particularly hard-working student). In some of these reactions, a displaced leaving group is also a good nucleophile. X R3C X Y X is a nucleophile Y is a leaving group CR3 Y X is a leaving group Y is a nucleophile Why is this reversibility an important issue? Imagine, for example, we needed to synthesize methyl iodide and chose to do this by the SN2 reaction of methyl chloride with iodide ion: CH3Cl + I CH3I + Cl This is potentially a reversible reaction because chloride ion reacts with methyl iodide to give methyl chloride and iodide ion, reducing the percentage of methyl chloride that is converted into methyl iodide. In general, can a leaving group function as a nucleophile and drive an SN2 reaction backwards? Is this equilibrium problem universal for SN2 reactions, and if so can it be controlled? Review Equilibrium and Le Châtelier’s principle (8.xx) Think Ahead Question 11.16 Suggest a reason why the SN2 reaction of CH3Cl with HO- has K eq > 1 whereas the SN2 reaction of CH3Cl with I- has Keq ~ 1. A protic solvent is used in both cases. Keq > 1 CH3Cl + HO CH3OH + Cl versus Keq ~ 1 CH3Cl + I CH3I + Cl Answer: Remember that every reaction and every mechanism step is an equilibrium, although we may not write it that way. For many reactions we study Keq is very large (products are highly favored). If Keq does not favor the products the reaction may not be useful to us unless we can manipulate or adjust the equilibrium somehow to make it more favorable. Ionic Substitution Reactions – Page 42 Remember also that an equilibrium favors the most thermodynamically stable set of molecules. When the reactants (species on the left of the equilibrium arrow) are more stable, the equilibrium favors the reactants over products and K eq < 1. When the products (species on the right of the equilibrium arrow) are more stable, the equilibrium favors the products over the reactants and Keq > 1. When the stability of the reactant and product sides of the equilibrium are about equal, neither side dominates and Keq~1. For many cases, including SN2 reactions, it may not be obvious just from looking at the reactants and products which side is more stable. We can view the equilibrium, however, as a pair of completing SN2 reactions. For example, the CH3Cl + HO- → CH3OH + Cl- reaction competes with CH3OH + Cl- → CH3Cl + HO-. We know that HO- is a good nucleophile and Cl- is a moderate leaving group, so CH3Cl + HO- → CH3OH + Cl- favors products. We also know that Clis a moderate nucleophile and HO- is a very bad leaving group so CH3OH + Cl→ CH3Cl + HO- lies very much to the left. Taken together, these two competing reactions cause the CH3Cl + HO- → CH3OH + Cl- equilibrium to lie to the right. CH3Cl + HO CH3OH + Cl poorer nucleophile better leaving group favors better nucleophile right side poorer leaving group In other words, the SN2 equilibrium favors the better leaving group and poorer nucleophile. Compare this with an acid/base equilibrium, which favors the weakest acid and weakest base (section 9.xx). In the CH3Cl + I- → CH3I + Cl- reaction, the better leaving group (I-) favors the left side of the equilibrium whereas the poorer nucleophile (Cl-) favors the right side of the equilibrium. There is no strong preference for either side of the equilibrium, so Keq ~1. CH3Cl + I better leaving group - favors left better nucleophile - favors right CH3I + Cl poorer leaving group - favors left poorer nucleophile - favors right Does this mean our methyl iodide synthesis is doomed to be inefficient, or can we manipulate the equilibrium? A common method to manipulate the position of an equilibrium takes advantage of Le Châtelier’s principle: when an equilibrium is disrupted by altering the concentration of one or more reactants, the equilibrium shifts to restore the original concentrations. For example, if the concentration of a product is reduced the equilibrium shifts to produce more of that product. If the concentration of a reactant is increased the equilibrium produces more product in order to remove the extra reactant. Both of these equilibrium perturbations are used in the laboratory to increase the amount of product produced. For example, SN2 reaction of an alkyl chloride with iodide ion (in order to produce an alkyl iodide) can be effectively shifted towards the alkyl iodide by using NaI as the nucleophile and acetone as the solvent. NaI is soluble in acetone Ionic Substitution Reactions – Page 43 Review An acid/base equilibrium favors the weakest acid and base (9.xx). whereas NaCl is much less soluble so it precipitates out as the reaction proceeds. (A precipitate is sometimes indicated with ↓ in a reaction equation.) This precipitation removes Cl- and continually shifts the equilibrium towards the product (Figure 11.15). The same procedure can also be used to convert an alkyl bromide into the corresponding iodide (NaBr precipitates). The reaction of an alkyl halide with NaI in acetone is called the Finkelstein reaction, named for Hans Finkelstein who first reported the reaction in 1910. R Cl + NaI (solvent = acetone) R X I + NaCl Figure 11.15: An alkyl chloride is converted to the corresponding alkyl iodide by SN2 reaction with NaI in acetone. The equilibrium is driven to the products by precipitation of NaCl. Concept Focus Question 11.48 For each SN2 reaction equilibrium decide if Keq > 1, Keq ~ 1 or Keq < 1. (a) CH3OSO2CF3 + Cl (b) CH3F + I CH3I + F Solvent = DMF (c) CH3F + I CH3I + F Solvent = CH3OH (d) PhCH2Br + PPh3 CH3Cl + PhCH2PPh3 Br OSO2CF3 Solvent = CH3OH Solvent = benzene 11.5 SN2 Reaction Analysis We have one last issue to address before our exploration of the SN2 reaction is complete. Just because we can write an SN2 reaction does not mean that it is practical. How can we analyze an SN2 reaction to determine if it occurs at a reasonable rate? Think Ahead Question 11.17 Based on what you have learned about the SN2 reaction in this chapter, prepare a checklist of features that an SN2 reaction needs to have in order to proceed at a reasonable rate. Answer: In this chapter we have identified four fundamental variables that influence the rate of an SN2 reaction: nucleophile, leaving group, steric hindrance and solvent. These form the basis of our SN2 Reaction Checklist. Ionic Substitution Reactions – Page 44 The SN2 Reaction Checklist Nucleophile: Must be moderate or better. Leaving group: Must be moderate or better. Steric hindrance: Carbon bearing leaving group cannot be 3o. Solvent: Polar aprotic preferred. Polar protic acceptable. Nonpolar rarely useful. If a reaction fails to meet all of these requirements it cannot proceed by the SN2 mechanism. (Other reaction mechanisms may occur, as we will learn later on. It is also possible that no reaction occurs at all). The requirements are somewhat interdependent. For example, a superior nucleophile will allow the reaction to proceed with a weaker leaving group, reduced steric hindrance allows the use of a poorer nucleophile, etc. Let’s see how the SN2 Reaction Checklist works by analyzing this reaction: I LiSCH3 DMF SCH3 • Nucleophile: CH3S- does not have resonance delocalization of electron density on sulfur. Sulfur has moderate atomic radius (3rd row element) and low electronegativity. There is no inductive effect to decrease electron density on sulfur. It has a formal negative charge. Therefore CH3S- is a good nucleophile. • Leaving group: Iodide is a superior leaving group, due mostly to its large atomic radius and high polarizability. • Steric hindrance: The carbon bearing the leaving group is secondary. This slows, but does not prevent an SN2 reaction. • Solvent: DMF is a polar aprotic solvent. • Conclusion: This SN2 reaction will proceed at a reasonable rate. Concept Focus Question 11.49 Will these SN2 reactions proceed at a reasonable rate? (a) CH3CH2Br NaOCH3 DMSO CH3CH2OCH3 Ionic Substitution Reactions – Page 45 (b) (CH3)3CI KCN (CH3)3CC DMF N O (c) LiF CH3CH2CH2O S CH3OH CH3CH2CH2F O (d) Cl H2O OH2 CH3CH2OH 11.6 The SN1 Reaction Back in section 11.2, we deduced the mechanism and transition state for the SN2 reaction. Some assumptions were made and later verified. One assumption, however, was made and not discussed: we assumed the reaction is concerted (all the bond making and bond breaking occurs in a single step). Now we will consider two alternate ionic substitution mechanisms. Review Reaction intermediate (8.xx). Think Ahead Question 11.18 Consider a stepwise ionic substitution mechanism for the generic reaction of R3C-LG (the electrophile) with a nucleophile (nuc) to give R3C-nuc. In this new mechanism the carbon-nucleophile bond is formed before the carbon-leaving group bond is broken, resulting in an intermediate structure. Draw the curved arrows for the mechanism, and suggest a very significant reason why ionic substitution cannot occur by this pathway. Answer: The mechanism involves two steps. In the first step, the nucleophile bonds to the carbon bearing the leaving group, to give an intermediate. In the second step, this intermediate decomposes to give R3C-nuc. R R C R R nuc LG R R nuc C LG R R C nuc + LG R Intermediate Carbon cannot have ten valence electrons under any circumstances. This mechanism is clearly impossible, because the intermediate has a carbon atom with ten electrons and five covalent bonds (a pentavalent carbon). Does an alternate timing of the bond change events lead to an acceptable mechanism? Think Ahead Question 11.19 Write an alternate mechanism for the reaction mentioned in Think Ahead Question 11.18, in which the carbon-leaving group bond breaks before the carbon-nucleophile bond is formed. This mechanism also includes an intermediate. Ionic Substitution Reactions – Page 46 Answer: The alternate mechanism features scission of the carbon-leaving group bond to give an intermediate. This intermediate then captures a nucleophile, resulting in a new carbon-nucleophile bond and the product. Writing out this mechanism reveals that the intermediate is a carbocation. (Unlike a pentavalent carbon, a carbocation is an acceptable reaction intermediate.) R R C LG R nuc C R R R Review R R C nuc + LG R Carbocation Let’s practice this new ionic substitution mechanism a few times before we explore it in detail. Concept Focus Question 11.50 Write a stepwise ionic substitution mechanism with a carbocation intermediate (not SN2) for the following reactions. O O (a) S O LiBr Br DMF CF3 I (b) (c) CH3SCH3 DMF (CH3)3CCl + H2O S(CH3)2 (CH3)3COH OCH3 I (d) CH3OH A. Kinetics Just because we can write a reaction mechanism does not mean the reaction follows that mechanism. In a broader sense, just because we can write a reaction does not mean that reaction occurs. Therefore we need to verify that ionic substitution can still occur by a mechanism other than SN2. In fact, there are many examples of ionic substitution reactions that occur readily, but cannot be SN2. One example is the reaction of tert-butyl chloride with 80% aqueous ethanol to give tert-butanol (Figure 11.16). This reaction cannot follow the SN2 mechanism because the carbon bearing the leaving group is tertiary. Ionic Substitution Reactions – Page 47 Carbocations (Chapter 10). This is a solvolysis reaction (section 11.3E) because water is both the solvent and nucleophile. Because water becomes part of the product, this is also a hydrolysis reaction. CH3 CH3 C CH3 CH3 Cl CH3 C OH2 CH3 CH3 CH3 Ionization of carbonleaving group bond forms a carbocation. C CH3 Carbocation captures nucleophile (a carbocation fate). H O CH3 OH2 CH3 H C OH CH3 Oxonium ion is deprotonated. Figure 11.16: Stepwise ionic substitution mechanism for the reaction of tertbutyl chloride with water to give tert-butanol. Review Rate-determining step (8.xx). Review Energy profile (8.xx). As we learned in section 11.2A, kinetic studies are useful to support a proposed mechanism. To do this we write a rate expression based upon the proposed mechanism, followed by kinetic studies in the lab to see if the proposed mechanism is consistent with the actual mechanism. This three-step mechanism leads to a rather complicated rate expression as long as none of the steps are ratedetermining (review section 8.xx if necessary). Think Ahead Question 11.20 Are any of the mechanism steps in Figure 11.16 rate determining? Draw an energy profile for the reaction. Answer: As we learned in our previous exploration of multistep mechanisms (section 8.xx), we can make an educated guess about the relative rate of mechanism steps by examining the bond changes in the transition state. This rough ΔH‡ analysis (Figure 11.17) is a good first step towards figuring out which mechanism step (if any) is rate determining. Recall from section 8.xx that endothermic mechanism steps (more bonds lost than gained) tend to have large energies of activation, whereas exothermic mechanism steps (more bonds formed than lost) and thermoneutral mechanism steps (same number of bonds lost and formed) tend to have small energies of activation. Also recall that the ratedetermining step has the largest energy of activation. Ionic Substitution Reactions – Page 48 Mechanism step CH3 CH3 C Cl CH3 CH3 CH3 CH3 C CH3 H O C-Cl none large C OH2 none C-OH small O-H O-H small CH3 CH3 OH2 CH3 H C CH3 OH2 CH3 ΔH‡ CH3 CH3 C Bonds made CH3 CH3 CH3 Bonds lost C OH + OH3 CH3 Figure 11.17: Looking for a rate-determining step in the hydrolysis of tert-butyl chloride. The analysis in Figure 11.17 leads us to conclude that the tert-butyl chloride hydrolysis mechanism has only one endothermic step: ionization of the carbonchlorine bond. By default this must also be the rate-determining step. (To rephrase the hourglass analogy we used in section 8.xx, the ionization of the carbon-chlorine bond is the narrowest of the three necks between the hourglass bulbs.) The mechanism steps in which the tert-butyl carbocation captures a molecule of water and the oxonium ion is deprotonated are much faster than the mechanism step in which the carbon-chlorine bond is ionized. Carbocation formation occurs in other ionic substitution reactions in this chapter as well as a variety of other organic mechanisms such as unimolecular βelimination (Chapter 12), addition to alkene and alkyne pi bonds (Chapter 13) and electrophilic aromatic substitution (Chapter 24). We will discover that in each of these mechanisms, carbocation formation is rate-determining. Therefore this is a good time to state a General Rule. General Rule In any reaction mechanism involving a carbocation, formation of the carbocation is the rate-determining step. With this information in hand, we can draw an energy profile for this reaction. The mechanism has three steps. Each mechanism step has its own transition state, so our energy profile has three humps. In addition, the mechanism includes two intermediates – a carbocation and an oxonium ion – so the energy profile has two valleys or saddle points. Ionic Substitution Reactions – Page 49 δ+ (CH3)3C δCl δ+ (CH3)3C δ+ OH2 (CH3)3CO δ+ Ea rds Energy H H OH2 δ+ (CH3)3C (CH3)3CCl (CH3)3COH2 (CH3)3COH Reaction coordinate Figure 11.18: Energy profile of the stepwise mechanism for the hydrolysis of tert-butyl chloride. After learning about the thermodynamics of these mechanism steps, students frequently ask, “if ionization of the carbon-leaving group bond is so energetically expensive, why does it happen in the first place?” In other words, why does the leaving group just get up and leave? To understand why, start by remembering that not all molecules in a reaction have the same energy. Molecules that do not have enough energy to achieve the transition state must somehow obtain more energy (perhaps by colliding with other molecules) before they can become product. Some of this energy is kinetic that causes bonds to vibrate. As the bonds vibrate the atoms move apart. If they have enough kinetic energy they can overcome the forces that hold the bond together and move away from each other (i.e., move through the transition state and break the bond). If the atoms do not have enough kinetic energy, then they move back together and another bond vibration cycle begins. If the bond is weak (stable leaving group, section 11.3B and/or stable carbocation, 11.3D) less energy is required to break the bond (energy of activation is lower). If the atoms have more kinetic energy (for example, because we have added heat to the reaction) then there is a greater chance the atoms have enough energy to move apart and break the bond. In summary, the bond “just breaks” because the atoms have enough energy to overcome the bonding forces, but stronger bonding forces require more energy to move the atoms apart. Now that we understand the reaction mechanism and a bit about its thermodynamics, we can deduce the rate law for the multistep ionic substitution reaction and give it a name. Review Relationship between rate determining step and rate expression (8.xx). Think Ahead Question 11.21 Write a rate expression for the hydrolysis of tert-butyl chloride. Remember the rate-determining step in the mechanism. Answer: For a multistep mechanism in which there is a rate-determining step, the rate expression is based only on that step (review section 8.xx if necessary). For the hydrolysis of tert-butyl chloride, the rate-determining step is also the first mechanism step: the ionization of the carbon-chlorine bond. This is a Ionic Substitution Reactions – Page 50 unimolecular (one molecule) mechanism step, so the rate expression is also based upon the concentration of just one molecule: Rate = k [(CH3)3CCl] Kinetic studies of other ionic substitution reactions at sp3 carbon that proceed through a carbocation intermediate show that this rate law is common to all. A generic rate law for these reactions is: Rate = k [R3C-LG] The rate law is first-order in the molecule undergoing substitution and first-order (unimolecular) overall. The concentration of the nucleophile does not enter into the rate expression. In contrast to their SN2 mechanism, Hughes and Ingold named this mechanism SN1 (substitution nucleophilic unimolecular). In a modern sense, any ionic substitution reaction at sp3 carbon that involves a carbocation intermediate is called SN1. Concept Focus Question 11.51 For the solvolysis reaction shown below, write the mechanism and product. Label the rate-determining step. Write the rate expression. O O S CH3 CH3OH O B. Stereochemistry Like the SN2 reaction, the SN1 mechanism has some consequences for the stereochemistry of the substitution product. Does an SN1 reaction give inversion of configuration like an SN2 reaction (section 11.2C) or is its stereochemical consequence different? Think Ahead Question 11.22 SN1 methanolysis (solvolysis in methanol) of (S)-1-phenethyl chloride gives 1phenethyl methyl ether. Is the product the R enantiomer, the S enantiomer or a mixture of the two? Hint: write the mechanism and identify the step(s) in which the stereochemistry of the product is established. Cl H OCH3 CH3 CH3OH Answer: Let’s start with the mechanism. Ionic Substitution Reactions – Page 51 CH3 H Cl Ph H H CH3 Ph H HOCH3 CH3 HOCH3 OCH3 Ph H CH3 (S) enantiomer Review Carbocation geometry (10.xx). OCH3 Ph CH3 mixture of enantiomers The mechanism reveals that the stereochemistry of the product is established when the carbocation captures methanol. The carbocation is planar (section 10.xx) and can be attacked on either face, giving both enantiomers of the oxonium ion intermediate. After proton transfer, the final reaction product consists of a mixture of ether enantiomers. Which enantiomer is the major one? Our mechanism predicts that both faces of the carbocation are attacked with equal probability. There is no bias for one face over the other. More rigorously stated, the transition states leading to the two oxonium ions are enantiomers. (Use a model of the carbocation and the oxonium ions to see this more clearly.) Recall that all physical properties of enantiomers (except the direction in which they rotate plane polarized light) are equal. Their energies of activation are equal, so they are formed at equal rates and in equal amounts – a racemic mixture. Do laboratory experiments support this prediction of a racemic product? Think Ahead Question 11.23 SN1 methanolysis of (S)-1-phenethyl chloride gives a 36:64 mixture of (S) and (R)-1-phenethyl methyl ether. What does this result suggest about the rate of attack on the two faces of the carbocation intermediate? Cl H CH3O CH3 CH3OH H H CH3 + S enantiomer (36%) OCH3 CH3 R enantiomer (64%) Answer: That the product mixture is not racemic means that one face of the carbocation was attacked more frequently than the other. The R enantiomer dominates, telling us that attack on the carbocation face opposite the departing leaving group is favored over attack on the same face as the departing leaving group. This bias makes no sense if the carbocation is naked (as depicted in the mechanism), but can be explained if the leaving group is still close to the carbocation when the nucleophile is captured. The leaving group provides some steric hindrance to attack, causing the nucleophile to favor (but not completely Ionic Substitution Reactions – Page 52 prefer) attack on the backside. In this case backside attack (leading to the R enantiomer and inversion of configuration) occurs about twice as often as front side attack (leading to the S enantiomer and retention of configuration). The association of a cation (the tert-butyl carbocation) and an anion (the leaving group) without forming a covalent bond is called an ion pair. Saul Winstein of UCLA pioneered the ion pair mechanism and its influence on ionic substitution reactions. Figure 11.19: Ionization of the carbon-chlorine bond forms a carbocation/chloride ion pair before the chloride ion diffuses away. The chloride ion hinders attack on one face of the carbocation, resulting in an unequal amount of the product stereoisomers. Studies of many other SN1 reactions have shown that this is a common phenomenon. General Rule An SN1 reaction produces both stereoisomers of the product at the carbon bearing the leaving group. Inversion is favored, but complete racemization is possible. Concept Focus Question 11.52 Give the SN1 solvolysis products for each reaction. Predict which stereoisomer is produced in the greatest amount. Cl H H (a) H2O H Cl (d) 80% CH3CH2OH 20% H2O Ph OTs I (b) CH2CH3 (e) Ph CH3OH H3 C H H CH3 D (c) Ph (CH3)3COH Ph OTs HOAc (f) OTf CH3 Ionic Substitution Reactions – Page 53 H2 O Saul Winstein (1912-1969), professor of chemistry at UCLA 1941-1969 and pioneer in the study of many areas of physical organic chemistry including ionic substitution reactions. 11.7 Factors Influencing the SN1 Reaction In section 11.3, we explored various factors (reaction variables) that might influence the rate and products of an SN2 reaction. Now let’s explore how these factors influence an SN1 reaction. A. The Nucleophile In section 11.3A we deduced that stronger nucleophiles make the SN2 reaction go faster. We also learned that the nucleophile determines the new functional group in the product. How do these properties of the nucleophile influence an SN1 reaction? Think Ahead Question 11.24 Select the fastest SN1 reaction. NaI Ph OTs DMF NaBr Ph I versus Ph OTs DMF Ph Br Answer: To understand the nucleophile’s influence on the reaction rate, we can examine the rate expressions. For SN2, rate = k [nucleophile][electrophile]. This rate expression reveals that there is a connection between the nucleophile and reaction rate. A change in the nucleophile (its concentration or nucleophilicity) will have an observable effect on reaction rate. For SN1, rate = k [R3C-LG]. This rate expression does not include the nucleophile, so any changes to the nucleophile will not have a significant impact on the reaction rate. In other words, the rate-determining step is perhaps millions of times slower than any other step in the SN1 mechanism. Changing the nucleophile does not change the rate-determining step and so the nucleophile’s impact on the reaction rate is insignificant. Therefore we predict that the SN1 reactions in question have nearly equal reaction rates. This is a very broad prediction and not without exceptions. For example, when the nucleophile is also the solvent its nature can have a profound impact on reaction rate (section 11.7D). The complete role of the nucleophile in the reaction must be considered before its effect can be fully ascertained. General Rule SN1 reaction rate is independent of the nucleophile, unless the nucleophile is also the solvent. The nucleophile also influences an SN1 reaction because it controls the identity of the new functional group in the product, exactly as it does for an SN2 reaction (section 11.3A). Ionic Substitution Reactions – Page 54 B. The Leaving Group In section 11.3B we saw that SN2 reaction rate is strongly dependent on the nature of the leaving group. Better leaving groups make for faster SN2 reactions. How does the leaving group influence SN1 reaction rate? Think Ahead Question 11.25 Select the fastest SN1 reaction. Ph I H2 O Ph versus OH Ph Cl H2 O Ph OH Answer: As we have seen several times before, reaction rate of a multistep mechanism is controlled by the rate-determining step. For an SN1 reaction, the rate-determining step is ionization of the carbon-leaving group bond to form the carbocation intermediate. A better leaving group will allow this ionization to proceed more readily, and the reaction as a whole will be faster. For the reactions in question, therefore, the hydrolysis of tert-butyl iodide is faster than the hydrolysis of tert-butyl chloride because iodide is a better leaving group than chloride. (Review leaving groups in section 11.3B if necessary.) General Rule SN1 and SN2 reaction rates both have the same dependence on leaving group: better leaving groups make for faster reactions. Concept Focus Question 11.53 For each reaction pair, select the faster SN1 reaction. Offer a brief explanation for each choice. LiF (a) Cl DMF I F LiF (b) Cl DMF (c) I DMF F DMF I LiI versus LiF (d) Cl F OH I DMF LiI versus DMF S(CH3)2 I LiF versus Ionic Substitution Reactions – Page 55 I LiI versus F DMF Cl DMF I Concept Focus Question 11.54 Select the major product and write a mechanism for its formation. Ph OTs Br + O CH3OH OH Br Ph Ph O Ph OTs Ph Ph Concept Focus Question 11.55 In the absence of other nucleophiles, some optically active molecules may racemize. Select the molecule that racemizes most rapidly and write a mechanism. Briefly explain your choice for the faster racemization. H Cl I H versus Ph Ph Concept Focus Question 11.56 2-Methyl-2-propanol does not react when treated with aqueous NaCl. Addition of H2SO4 to the reaction mixture results in rapid production of 2-chloro-2methylpropane. (a) Write a mechanism for the reaction with H2SO4 present. (b) Explain why the alcohol does not react until acid is added. C. Carbocation Stability In section 11.3D we discovered that the rate of an SN2 reaction is sensitive to steric hindrance: increasing the number of substituents on the carbon bearing the leaving group causes the reaction to be slower. How does the degree of substitution at this same carbon influence an SN1 reaction? Think Ahead Question 11.26 Rationalize the following relative rate data for the SN1 hydrolysis of several alkyl bromides. RBr H2O ROH RBr CH3Br CH3CH2Br (CH3)2CHBr (CH3)3CBr krel <1 1.0 12 1.2 x 106 Answer: When discussing the relative rate of SN1 reactions, we focus on the ratedetermining step of the mechanism, which is ionization of the carbon-bromine bond to form a carbocation: R3C-LG → R3C+ + LG. The molecules in question differ by the number of methyl groups attached to the carbon bearing the leaving group. So we can rephrase the question: how does the number of methyl groups on the carbon bearing the leaving group influence the rate of ionization? Ionic Substitution Reactions – Page 56 The rate of ionization is controlled by the energy of activation, which in turn in controlled by the energy difference between the reactant (RBr) and the transition state. This is a highly endergonic mechanism step, so according to Hammond’s postulate (section 8.xx) the transition state has a significant amount of carbocation character. Therefore the carbocation stability has strong influence on the energy of activation. The greater the stability of the carbocation intermediate, the lower the energy of activation for its formation. In other words, more stable carbocations are formed more quickly. R C Energy Ea 2 o R H Br R C R H Energy Reaction coordinate R Ea 3 o Br R C C R R R R Reaction coordinate + Figure 11.20: The effect of R3C stability on R3C-Br ionization rate. More stable carbocations have lower energy of activation and are formed more quickly. This prediction is in good agreement with the RBr SN1 hydrolysis rate data. Hydrolysis is faster when the carbocation intermediate is more stable. SN1 hydrolysis rate CH3Br < CH3CH2Br < (CH3)2CHBr < (CH3)3CBr Carbocation stability CH3+ < CH3CH2+ < (CH3)2CH+ < (CH3)3C+ Carbocation type Methyl 1o 2o 3o The results of many SN1 rate studies have shown that the rate dependence of carbocation stability is a common feature to all SN1 reactions. Ionic Substitution Reactions – Page 57 Review Hammond’s postulate (8.xx). Relative stability of carbocations (10.xx). General Rule SN1 reaction rate parallels carbocation stability: R3C-LG (tertiary) > R2CH-LG (secondary) > RCH2-LG (primary) >> CH3-LG (methyl). In addition, an SN1 reaction involving a resonance-stabilized carbocation intermediate is faster than a reaction involving a similar carbocation without resonance. Review Methyl carbocation is highly unstable (section 10.xx). The methyl carbocation (+CH3) is so highly unstable (section 10.xx) that SN1 reactions that would involve it cannot happen (unless it is a resonance-stabilized methyl carbocation such as +CH2OCH3). Increasing steric hindrance to nucleophilic attack SN2 favored CH3-LG RCH2-LG R2CH-LG R3C-LG SN1 favored Decreasing carbocation stability Figure 11.21: Effect on ionic substitution reaction rate versus number of substituents on the carbon bearing the leaving group. Concept Focus Question 11.57 For each pair of molecules select the one that undergoes the fastest SN1 reaction. (a) 1-iodopropane versus 2-iodopropane (b) 1-iodopropane versus allyl iodide (c) 2-iodo-3-butene versus 3-iodo-1,4-pentadiene I (d) I versus Ionic Substitution Reactions – Page 58 Concept Focus Question 11.58 Write the mechanism for the faster of these two methanolysis reactions. CH3OH OTs OCH3 versus OCH3 OTs CH3OH D. Solvent Effects In section 11.3E, we discovered that the nature of the solvent plays a critical role in the rate of an SN2 reaction. For example, the SN2 reaction of CH3Br with F- in DMF (a polar aprotic solvent) is very fast but in CH3OH (a polar protic solvent) it is very slow. What is the role and effect of solvent in an SN1 reaction? Think Ahead Question 11.27 In order to ascertain the effect of solvent on an SN1 reaction, the rate of tert-butyl chloride solvolysis in several solvents (ROH) was explored. What do the results suggest about the relationship between solvent dielectric constant and SN1 reaction rate? Offer an explanation for this relationship. (CH3)3C-Cl ROH (CH3)3C-OR ROH CH3CH2OH 40% H2O 60% CH3CH2OH* 80% H2O 20% CH3CH2OH H2O krel 1 100 14,000 100,000 *A solvent mixture prepared with 4 mL H2O for every 6 mL CH3CH2OH. Answer: As in previous sections where we explored the effect of various factors on the rate of an SN1 reaction, here we focus on the effect of the solvent on the SN1 rate-determining step. Let’s rephrase the question: why does increasing the percentage of water in the solvent cause faster ionization? One solvent property that can influence the rate of ionization is the dielectric constant (ε), the solvent’s ability to insulate or separate unlike charges. (Review dielectric constant in section 11.3E if necessary.) As the carbon-chlorine bond of tert-butyl chloride ionizes, unlike charges are forming: a positively charged tertbutyl carbocation and a negative charged chloride ion. The more effective the Ionic Substitution Reactions – Page 59 solvent is at separating ions (i.e., the higher its dielectric constant) the less energy will be required for the ionization. In other words ionization is faster in a more polar solvent. Ethanol has ε = 25, water has ε = 80 and mixtures of these solvents have ε values somewhere in between. Therefore this SN1 reaction is faster in pure water than in pure ethanol because water is more polar. Here is a more rigorous explanation. The ionization rate is a function of the energy of activation, the energy difference between tert-butyl chloride and the corresponding ions. The transition state has a significant level of charge on carbon and chlorine whereas the reactants have much less. A polar solvent stabilizes this polar transition state more effectively than it does the neutral reactants. More polar solvents give more stabilization, and a correspondingly larger decrease in the energy of activation. Energy CH3CH2OH 40% H2O/60% CH3CH2OH H2O (CH3)3C+ + Cl- (CH3)3CCl Reaction coordinate Figure 11.22: A polar solvent stabilizes the ionization transition state more than the reactants. Energies of activation: CH3CH2OH > 40% CH3CH2OH/60% H2O > H2O. Increasing solvent polarity results in lower energy of activation and faster ionization. Studies on many other SN1 reactions show that this relationship between solvent polarity and reaction rate is general. General Rule Increasing solvent polarity increases SN1 reaction rate. Note that both SN2 (section 11.3E) and SN1 reactions are best in polar solvents. However, the reasons for this are quite different. Polar solvents are usually used in SN2 reactions in order to dissolve the anionic nucleophile whereas a polar solvent is best for an SN1 reaction because it enhances ionization of the carbonleaving group bond. Proticity (the ability to donate a hydrogen for hydrogen bonding) is another important solvent characteristic. In section 11.3E we learned that SN2 reactions are slower in protic solvents than in aprotic solvents because hydrogen bonding Ionic Substitution Reactions – Page 60 from protic solvents decreases nucleophilicity (the “paparazzi effect”). How does proticity influence the SN1 reaction? Hydrogen bonding assists the ionization of the carbon-leaving group bond by stabilizing the leaving group as it begins to leave. This stabilization lowers E a and accelerates the ionization. (Compare this with the way in which hydrogen bonding stabilizes a nucleophile, and decreases its reactivity.) Most SN1 reactions are conducted in polar, protic solvents because both of these solvent features assist the SN1 rate-determining step and help make the reaction faster. Concept Focus Question 11.59 Select the faster SN1 reaction in each pair. Write the mechanism and product(s) for the faster reaction. I (a) CH3OH OTs (b) (c) I versus CH3CH2OH OTs 90% ethanol 10% water versus 10% ethanol 90% water I I CH3CH2OH versus CH3OH Concept Focus Question 11.60 The relative rate of solvolysis of tert-butyl bromide in ethanol, water and formic acid was found to be ethanol < formic acid < water. Based upon these relative rates, assign the following ε values to the solvents used in this study: 80, 58 and 25. (Try to do this without referring to the solvent properties table earlier in this chapter.) Concept Focus Question 11.61 Figure 11.22 shows that as the SN1 reaction solvent becomes more polar, the ratedetermining transition state becomes earlier. Explain why this is so in terms of Hammond’s postulate. Concept Focus Question 11.62 (a) Make a drawing of the transition state that shows how CH3OH assists carbonleaving group bond ionization, and thus accelerates the SN1 methanolysis of (CH3)3CBr. Hint: CH3OH is a polar, protic solvent. (b) Which ionization is faster in CH3OH: (CH3)3C-Cl or (CH3)3C-S(CH3)2+? Remember to consider solvent effects on both the reactants and the transition state. Ionic Substitution Reactions – Page 61 11.8 SN1 Reaction Analysis We have one last issue to address before our exploration of the SN1 reaction is complete. Just because we can write an SN1 reaction does not mean that it is practical. How can we analyze an SN1 reaction to determine if it occurs at a reasonable rate? Think Ahead Question 11.28 Review our SN2 Reaction Checklist in section 11.5. Based on what you have learned about the SN1 reaction in this chapter, prepare a checklist of features that an SN1 reaction needs to have to proceed at a reasonable rate. Answer: In this chapter we have identified three basic variables that influence the rate of an SN1 reaction: leaving group, carbocation stability and solvent. (Remember that the nucleophile has no significant effect on SN1 reaction rate because it is not involved in the rate-determining mechanism step.) These form the basis of our SN1 Reaction Checklist. The SN1 Reaction Checklist Leaving group: Must be moderate or better. Carbocation stability: Carbocation must be secondary or better. Solvent: Polar is necessary; protic is preferred. If a reaction fails to meet all of these requirements it cannot proceed by the SN1 mechanism. Instead, there might be no reaction or it might proceed by the SN2 mechanism. (The issue of SN2 versus SN1 will be addressed in section 11.9.) The requirements are somewhat interdependent. For example, a superior leaving group (such as iodide or tosylate) or more stable carbocation will allow the reaction to proceed with a less polar solvent. Let’s see how the SN1 Reaction Checklist works by analyzing this reaction: O O S OCH3 O CH3OH CF3 • Leaving group: The leaving group is CF3SO3- (trifluoromethanesulfonate). We learned in section 11.3B that sulfonates are superior leaving groups due to their significant resonance and inductive stabilization. Therefore the leaving group requirement is met very well. Ionic Substitution Reactions – Page 62 • Carbocation stability: The carbocation formed after ionization of the leaving group is tertiary with resonance when we consider the best resonance contributor. O O S O CF3 This carbocation has very good stability (at least for a carbocation) and so the carbocation requirement is met very well. • Solvent: Methanol is a polar (ε = 33) protic solvent, which favors ionization of the carbon-leaving group bond. The solvent requirement is met very well. The SN1 requirements are met very well, so we predict this SN1 reaction will proceed at a very good rate. Methanol is a poor nucleophile but this is inconsequential because the SN1 ratedetermining step does not include the nucleophile. Concept Focus Question 11.63 Decide if each of the following is a reasonable SN1 reaction. OCH2CH3 Br (a) CH3O CH3CH2OH (b) (c) F 80% H2O 20% CH3CH2OH CF3CH2OTs I (d) CH3OH OH CF3CH2OCH3 Cl NaCl THF OH Cl (e) CH3O 95% acetone 5% H2O Concept Focus Question 11.64 The following reactions cannot occur by the SN1 mechanism. For each reaction, state why it cannot follow the SN1 mechanism. Also, by adding, subtracting or otherwise changing at most four atoms rewrite the reaction so that it can readily occur by the SN1 mechanism. Ionic Substitution Reactions – Page 63 NH2 (a) (b) CH3OH CH3CH2OTs I (c) OCH3 80% CH3OH 20% H2O LiBr CH3CH2OCH3 Br CH2Cl2 11.9 Competition Between SN2 and SN1 Mechanisms So far in this chapter we have treated SN2 and SN1 as two distinctly different mechanisms. You probably have noticed a number of similarities between these mechanisms, the most obvious similarity is that they both start with a carbonleaving group bond and end up with a carbon-nucleophile bond. (You may find it useful to prepare a more complete list of the similarities and differences between the SN2 and SN1 reactions.) In some cases an ionic substitution reaction obviously follows the SN2 mechanism (for example when the electrophile is methyl bromide) or the SN1 mechanism (for example when the electrophile is tert-butyl bromide). What mechanism occurs when both the SN2 and SN1 are possible (perhaps with a secondary electrophile such as 2-iodopropane)? Does some sort of SN2/SN1 hybrid mechanism occur, or is there a preference for SN2 versus SN1? There are two common areas of thought among chemists on this issue. In some cases, there is evidence for an SN2/SN1 hybrid mechanism. This hybrid mechanism combines features of both SN2 and SN1. For example, when the nucleophile attacks the carbon bearing the leaving group, the carbon-leaving group bond is already elongated (partially broken) but the leaving group has not fully ionized away to leave a carbocation. This hybrid mechanism a natural extension of the Winstein ion pair mechanism we encountered back in section 11.6B. In other cases, the evidence suggests that the ionic substitution mechanisms compete. Some molecules may react by an SN2 mechanism while others in the same flask follow the SN1 mechanism. In this case how do we know which mechanism dominates? (As it turns out, this will also help us decide if the hybrid mechanism has more SN2 or more SN1 character.) Ionic Substitution Reactions – Page 64 Think Ahead Question 11.29 The ionic substitution reaction shown below might occur by either the SN2 or SN1 mechanism. Predict which mechanism dominates. OTs I NaI CH3OH Answer: This is a borderline mechanism case because it can occur readily by either the SN2 or SN1 mechanism. SN2 is favored by the excellent leaving group and good nucleophile. SN1 is favored by the excellent leaving group, excellent carbocation and polar protic solvent. So which mechanism dominates? Put yourself in Mother Nature’s shoes (so to speak). When given a task to complete (ionic substitution) and two ways to achieve the task (SN2 or SN1) which do you pick? Generally you pick the easiest way to get the job done. Reactions do the same thing: pick the pathway of least resistance. So now let’s ask a new question: between SN2 and SN1, which requires the least amount of energy? Everything else being equal, SN2 avoids the energetically expensive carbocation required by SN1. In other words, the rate-determining step for SN2 is often lower energy than the rate-determining step for an SN1 mechanism that achieves the same reaction. Therefore for a borderline mechanism case, we consider SN2 before SN1. If the SN2 requirements are met the SN2 mechanism will dominate. If the SN2 conditions are not met and the SN1 conditions are met the SN1 mechanism dominates. If neither set of conditions is met the reaction will probably not occur by either the SN2 or SN1 mechanism. This is a useful strategy to analyze any ionic substitution reaction at sp3 carbon. So for the reaction of Think Ahead Question 11.29 the dominant mechanism is SN2. Here is an alternate viewpoint. Because the SN2 rate-determining step is generally lower energy than the corresponding SN1 rate-determining step, at any given time more molecules have enough energy to cross the SN2 rate determining transition state than to cross the SN1 rate determining transition state. Therefore a greater number of molecules follow the SN2 mechanism than follow the SN1 mechanism. (Imagine you have to get out of a valley that is flanked by mountains on either side, but one set of mountains is a bit lower than the other. Which mountain range do you cross? Think of these mountain ranges as transition states as shown in Figure 11.23.) In practice, the energy difference between the SN2 and SN1 mechanisms may be very small. A very subtle factor, easily overlooked, might tip the balance in favor of either mechanism. Ionic Substitution Reactions – Page 65 SN2 Mountain SN1 Mountain Valley of the Reactants Figure 11.23: Which mountain do you climb to escape the Valley of the Reactants? Concept Focus Question 11.65 Decide if the following reactions occur mostly by the SN2 mechanism, the SN1 mechanism, or not at all. Give the product and write out the most probable mechanism. (a) CH3CH2CH2CH2Br S DMF (CH3CH2)3N (d) Cl acetone O I (b) CH3OH (e) OTs Ph F (c) THF H CF3CO2 CH2Cl2 Concept Focus Question 11.66 By adding, subtracting or otherwise changing at most six atoms in each, rewrite reactions (a) – (c) of Concept Focus Question 11.65 so that they proceed by the mechanism that you have not chosen. For example if you decided the reaction follows the SN2 mechanism, change it so it follows the SN1 mechanism. Concept Focus Question 11.67 Many factors can shift the delicate balance between SN2 and SN1 mechanisms. Here are two of these factors. Fill in each blank with SN2 or SN1. (a) A good nucleophile favors ____ whereas a weak nucleophile favors ____. (b) A high concentration of nucleophile favors ____ whereas a low concentration of nucleophile favors ____. 11.10 In the Real World: S-Adenosylmethionine A living cell is a very complex chemical machine, but the reactions that occur there are subject to the same rules and limitations as any other organic reactions. Therefore we can begin to understand these reactions by considering their similarities to reactions that we have already studied (or will study later on). Ionic Substitution Reactions – Page 66 There are many examples of ionic substitution reactions among the plethora of reactions that occur in a cell. One of these is the transfer of a methyl group from one molecule to another. What is the mechanism of these methyl transfer reactions? What biomolecules provide the methyl groups (i.e., are the electrophiles) and what biomolecules accept the methyl groups (i.e., are the nucleophiles)? Any methyl transfer mechanism must be an SN2 process, because a methyl carbocation is much too unstable to exist under normal (especially biological) conditions. S-adenosylmethionine (SAM) is one of several biological methylating agents. (You can think of this molecule as “Nature’s iodomethane.”) It works in conjunction with a group of enzymes called methyl transferases that mediate the methyl transfer. NH2 This methyl group is transferred N O N CH3 S N O The S of S-adenosylmethionine means the adenosine group is attached to methionine at sulfur, just like the N,N designation in N,N-dimethylformamide. It is not an indication of stereochemical configuration. N O NH3 HO OH S-adenosylmethionine (SAM) SAM is involved in a wide range of biological methylations, most of which occur in the liver. For example it provides a methyl group to convert norepinephrine into epinephrine (adrenaline). Norepinephrine and epinephrine are hormones released into the bloodstream in response to stress. OH OH HO SAM HO NH2 N HO HO Norepinephrine H CH3 Epinephrine (adrenaline) SAM is required for the conversion of phosphatidyl ethanolamine (an important cell membrane component) into phosphatidyl choline (lecithin). Lecithin is another important cell membrane component that protects cells from oxidation and largely comprises the protective sheaths surrounding the brain. (These molecules are lipids, which you can learn more about in section 29.xx.) O O 3 SAM RO2C O RO2C P CH2CH2NH2 O Phosphatidyl ethanolamine RO2C O RO2C P CH2CH2N(CH3)3 O Phosphatidyl choline (lecithin) Ionic Substitution Reactions – Page 67 SAM methylates guanidoacetate to produce creatine, which is used for storage of high-energy phosphate. NH2 NH2 SAM H2N N CO2 H2N N H CO2 CH3 Guanidoacetate Creatine Although these examples focus in reactions in which SAM methylates nitrogen atoms, it methylates sulfur and carbon in other biosynthetic pathways. Concept Focus Question 11.68 Draw the structure of the leaving group formed when SAM gives up its methyl group. Why is this a good leaving group? Concept Focus Question 11.69 Why does SAM methylate norepinephrine at the amine instead of any of the OH groups? Concept Focus Question 11.70 (a) Does the conversion of guanidoacetate into creatine by SAM occur by an SN2 or an SN1 mechanism? (b) Write the mechanism. Assume the enzyme that mediates this process (abbreviated enz-NH2) can also function as a base. Concept Focus Question 11.71 Guanidoacetate has four potentially nucleophilic sites for methylation: three nitrogen atoms and the carboxylate group. (Assume the enzyme enz-NH2 is available to act as a base if needed, but only after the new methyl group has been installed.) Does SAM methylate the most nucleophilic site of guanidoacetate? Concept Focus Question 11.72 Upon accepting a proton the molecule shown below becomes N5-methyltetrahydrofolate (MTH), a methyl transfer molecule like SAM. Does the protonation occur at N5 or at N8? H H2N N N8 N H N N H N R N5 O CH3 Ionic Substitution Reactions – Page 68 Chapter Summary 11.1 Why Should I Study This? An ionic substitution reaction involves replacement of a leaving group by a nucleophile at an sp3 carbon. It is a fundamental reaction in organic synthesis and biosynthesis. 11.2 The SN2 Reaction In the concerted ionic substitution mechanism, the carbon-nucleophile bond is formed at the same time the carbon-leaving group bond is broken. The nucleophile determines the nature of the new functional group in the product. R1 R1 Nuc C R2 R3 LG Nuc R1 C R3 LG R2 Nuc + LG C R2 R3 A. Mechanism and Kinetics The rate expression for this concerted ionic substitution is: rate = k [nucleophile] [electrophile]. Based on this rate expression, the mechanism is called SN2 (substitution nucleophilic bimolecular). B. Transition State The SN2 transition state features formation of the carbon-nucleophile bond simultaneously with the rupture of the carbon-leaving group bond. C. Inversion of Configuration The nucleophile attacks the carbon-leaving group σ* orbital at the backside of the carbon-leaving group bond, leading to inversion of configuration. In general, an R stereocenter is inverted to S, and S becomes R. 11.3 Factors Influencing the SN2 Reaction The rate and product of an SN2 reaction is influenced by the nucleophile, the leaving group, steric hindrance and the solvent. A. The Nucleophile The role of the nucleophile is to share an electron pair as the carbonnucleophile bond is forming. A stronger nucleophile is more willing to share an electron pair, and causes a faster SN2 reaction. There is a good parallel between basicity and nucleophilicity, and the factors that influence them. These factors include resonance, electronegativity, atomic radius, inductive effects and formal charge. B. The Leaving Group The role of the leaving group is to accept an electron pair as the carbonleaving group bond breaks. A better leaving group can accommodate the electron pair more effectively. In general the same factors that influence nucleophilicity also influence leaving group ability, but in an opposite sense. Ionic Substitution Reactions – Page 69 C. A Few Words About Polarizability Polarizability is the ability of an electron cloud to be distorted in response. The electron cloud can be soft (easily distorted) or hard (not easily distorted). In SN2 reactions, higher polarizability enhances nucleophilicity and leaving group propensity. D. Steric Effects Increasing the number and size of substituents on the carbon bearing the leaving group increases the van der Waals repulsions in the SN2 transition state, causing a slower reaction. CH3-LG (methyl; fastest) > RCH2-LG (1o) > R2CH-LG (2o) > R3C-LG (3o; slowest) E. Solvent Effects Solvent effects on reactions are based their ability to insulate unlike charges (dielectric constant, ε) and hydrogen bonding (proticity). A protic solvent can serve as a hydrogen bond donor (usually with an O-H), whereas an aprotic solvent is not a hydrogen bond donor. The best solvent for most SN2 reactions is polar (to dissolve the anionic nucleophile) and aprotic (to avoid hydrogen bonding which decreases nucleophilicity). 11.4 Reversibility of the SN2 Reaction An SN2 reaction is an equilibrium, which generally favors the best leaving group and weakest nucleophile. Le Châtelier’s principle can be used to manipulate an unfavorable equilibrium by, for example, precipitation of a reaction product (the Finkelstein reaction). 11.5 SN2 Reaction Analysis In order for an SN2 reaction to proceed at a reasonable rate it needs a moderate or better nucleophile, a moderate or better leaving group, the carbon bearing the leaving group cannot be tertiary, and the solvent usually polar aprotic. 11.6 The SN1 Reaction An alternate, stepwise ionic substitution mechanism is possible. This mechanism features two steps: ionization of the carbon-leaving group bond to form a carbocation (a reaction intermediate), following by capture of a nucleophile by the carbocation. The nucleophile is frequently a solvent molecule, in which case the reaction is called solvolysis. R1 R1 R1 C R2 R3 LG Nuc C R3 R2 R2 C Nuc R3 A. Kinetics The slowest step in this stepwise ionic substitution mechanism is the ionization of the carbon-leaving group bond. The rate law includes only the molecule suffering ionization, and not the nucleophile: Ionic Substitution Reactions – Page 70 rate = k [R3C-LG]. Based on this rate law the mechanism is called SN1 (substitution nucleophilic unimolecular). B. Stereochemistry The carbocation may capture a nucleophile from either face, so the product is a mixture of stereoisomers. More of the inversion product is formed than the retention product because the leaving group provides some steric hindrance to nucleophilic attack on one face of the carbocation. 11.7 Factors Influencing the SN1 Reaction The rate and product of an SN1 reaction are influenced by the nucleophile, the leaving group, carbocation stability and solvent. A. The Nucleophile The nucleophile influences the product, but not the rate of an SN1 reaction. B. The Leaving Group Better leaving groups make for faster SN1 reactions. The factors that influence leaving group ability are independent of reaction mechanism. C. Carbocation Stability Formation of the carbocation is the SN1 rate-determining step, so a more stable carbocation intermediate makes for a faster SN1 reaction. CH3-LG (slowest) < RCH2-LG < R2CH-LG < R3C-LG (fastest). SN1 reactions involving resonance-stabilized carbocations are more stable than SN1 reactions involving similar carbocations without resonance stabilization. D. Solvent Effects An SN1 reaction is fastest in a polar protic solvent because this type of assists ionization most effectively. 11.8 SN1 Reaction Analysis In order for an SN1 reaction to proceed at a reasonable rate it needs a moderate or better leaving group, a carbocation intermediate which is at least secondary or primary with resonance, and a polar solvent. 11.9 Competition Between SN2 and SN1 Mechanisms When reaction conditions allow for either SN2 or SN1, SN2 generally dominates because it is an energetically less expensive pathway. It can also be said that a hybrid SN2/SN1 mechanism operates. 11.10 In the Real World: S-Adenosylmethionine SN2 reactions are important in biosynthetic pathways. S-Adenosylmethionine is an important biochemical electrophile that provides a methyl group in various biochemical pathways. Ionic Substitution Reactions – Page 71 New Terms Ionic substitution reaction (page 1) Organic synthesis (page 1) SN2 (substitution nucleophilic bimolecular) (page 5) Backside attack (page 7) Inversion of configuration (Walden inversion) (page 10) Retention of configuration (page 10) Ambident nucleophile (page 17) Sulfonate (page 27) Polarizability (page 29) Methyl carbon (page 31) Primary (1o) carbon (page 31) Secondary (2o) carbon (page 31) Tertiary (3o) carbon (page 31) Quaternary (4o) carbon (page 31) Steric hindrance (page 32) Solvent (page 34) Solvolysis (page 34) Proticity (page 34) Dielectric constant, ε (page 34) Protic solvent (page 36) Aprotic solvent (page 36) Polar solvent (page 36) Nonpolar solvent (page 36) Solvation (page 37) Solvent shell (page 37) Finkelstein reaction (page 44) SN1 (substitution nucleophilic unimolecular) (page 51) Ion pair mechanism (page 53) S-adenosylmethionine (SAM) (page 66) Ionic Substitution Reactions – Page 72 Concept Review Questions These concept review questions refer this reaction: H3C C H I NaSCH3 CH3OH 1. Write the product and mechanism for this SN2 reaction. Include all transition states. 2. Write the SN2 rate expression for this reaction. 3. Use this reaction as a basis to explain “inversion of configuration.” 4. Briefly discuss how each of the following factors influences nucleophilicity: resonance, electronegativity, atomic radius, inductive effect and formal charge. What is the sequence of relative influence of these factors? 5. (a) Explain why iodide ion is a better leaving group than chloride ion or hydroxide ion. (b) What structural features make trifluoromethanesulfonate ion a superior leaving group? 6. What is polarizability, and how does it influence nucleophilicity and leaving group propensity? 7. Is the SN2 reaction of methyl iodide with NaSCH3 in methanol faster or slower the reaction above? Explain. 8. (a) If the solvent is changed to DMF, is the SN2 reaction faster or slower? Explain. (b) If the solvent is changed to ethanol, is the SN2 reaction faster or slower? Explain. 9. Some SN2 reactions are easily reversible but this one is not. Explain why Keq is large in this case. 10. Does this SN2 reaction proceed at a reasonable rate? 11. Write the SN1 solvolysis mechanism for this reaction. Label the rate-determining step. 12. Predict the effect on the SN1 reaction rate when NaSCH3 is changed to NaBr. 13. Predict the effect on the SN1 reaction rate when the leaving group is changed to chloride. Ionic Substitution Reactions – Page 73 14. Predict the effect on the SN1 reaction rate when the methyl group of the electrophile is changed to a hydrogen atom. 15. Predict the effect on the SN1 reaction when methanol is changed to ethanol. 16. Does the SN1 reaction (as shown above) proceed at a reasonable rate? 17. Does the reaction shown above occur by the SN2 or the SN1 mechanism? Ionic Substitution Reactions – Page 74