Review
TRENDS in Biochemical Sciences
Vol.30 No.11 November 2005
Mammalian cyclin-dependent kinases
Marcos Malumbres and Mariano Barbacid
Molecular Oncology Programme, Centro Nacional de Investigaciones Oncológicas (CNIO), Melchor Fernández Almagro 3, E-28029
Madrid, Spain
Cyclin-dependent kinases (Cdks) are the catalytic subunits of a family of mammalian heterodimeric serine/
threonine kinases that have been implicated in the
control of cell-cycle progression, transcription and
neuronal function. Recent genetic evidence obtained
with gene-targeted mice has shown that Cdk4 and Cdk6
are not needed for entry into the cell cycle after
mitogenic stimuli and organogenesis; however, they
are essential for the proliferation of some endocrine and
hematopoietic cells. Cdk2 is also dispensable for the
mitotic cell cycle. Indeed, mice without Cdk2 are normal
except for their complete sterility: unexpectedly, Cdk2 is
crucial for the first meiotic division of male and female
germ cells. These findings have important implications
both for our current understanding of the role of Cdks in
regulating the mammalian cell cycle and for their
potential use as therapeutic targets in cancer.
Introduction
Cdks are the catalytic subunits of a large family of
heterodimeric serine/threonine protein kinases whose
best-characterized members are involved in controlling
progression through the cell cycle. According to the latest
versions of the human and mouse genomes, there are 11
genes encoding Cdks and 9 other genes encoding Cdk-like
proteins with conserved primary structure (Figure 1).
Because the catalytic activity of these Cdks requires
binding of a regulatory subunit, the term Cdk is often used
for the active heterodimeric complex. In this review, we
have made every effort to avoid this confusing terminology. The activating partners of the cell-cycle Cdks are
molecules that are synthesized and degraded during each
cell cycle and thus have been designated ‘cyclins’ (see
later). Although this property has been used to define this
kinase family, not all activating partners of Cdks are
synthesized and destroyed in a cyclical fashion. Indeed,
the physiological role of most Cdks and their activating
partners remains unknown.
In this review, we first summarize the most relevant
biochemical information for known Cdks, with a particular emphasis on those involved in regulating the cell cycle.
We then discuss this information in view of recent
observations derived from genetic studies in mice that
have challenged some of the widely accepted models of the
mammalian cell cycle. Finally, we consider the implications of these findings for the use of cell-cycle Cdks as
Corresponding authors: Malumbres, M. (malumbres@cnio.es), Barbacid, M.
(barbacid@cnio.es).
targets for the development of selective inhibitors with
potential therapeutic value in cancer.
Nomenclature: a historical account
The original member of the Cdk family (now designated
Cdk1) was identified in genetic screens for Schizosaccharomyces pombe and Saccharomyces cerevisiae mutants
with defects in the cell division cycle [1]. This protein,
designated Cdc2 in S. pombe and Cdc28 in S. cerevisiae,
was shown to be essential for cell-cycle progression. Soon
after, homologs of Cdc2 were identified in human cells by
their ability to complement yeast mutants [2,3]. Using an
independent approach, Hanks [4] cloned a related gene
(termed PSK-J3 for putative serine/threonine kinase,
filter J colony 3) by hybridizing a HeLa cell cDNA library
with oligonucleotide probes homologous to known serine/
threonine kinases. Subsequently, another member of the
Cdk family, designated Cdk2, was cloned by three
independent approaches based on the complementation
of S. cerevisiae cdc28 mutants, differential display or
interaction with Cyclin A [5–8]. The advent of PCR
technology led to the identification of additional family
members by using degenerate primers to amplify human
cDNAs [9–12]. Some of these new members were
designated PSSALRE or PLSTIRE on the basis of their
amino acid sequence in a conserved domain.
To provide a unifying nomenclature for this emerging
gene family, scientists decided to adopt the term ‘cyclindependent kinases’ during the Cold Spring Harbor
Symposium on Cell Cycle in 1991. As a consequence,
Cdc2 became Cdk1, PSK-J3 was renamed Cdk4, and
PSSALRE and PLSTIRE became Cdk5 and Cdk6,
respectively. Additional members of the mammalian Cdk
family that have been cloned and characterized in
subsequent years include Cdk7 [13], Cdk8 [14], Cdk9
[15], Cdk10 [16] and Cdk11 [17]. Other highly related
molecules, such as members of the PCTAIRE or PFTAIRE
group of proteins, as well as CCRK, CHED and CRK7,
have not been formally designated ‘Cdks’ because no
activating subunit has been identified for these molecules
(Table 1, Figure 1).
Independent studies using sea urchin eggs identified a
series of proteins that were synthesized and destroyed at
each cleavage division. On the basis of this feature, these
proteins were designated ‘cyclins’ [18]. Cyclins were
subsequently cloned from fertilized clam and sea urchin
embryos and shown to promote meiosis in Xenopus laevis
oocytes [19]. Related proteins, designated Cdc13, Cln and
Cyclin B, were isolated from S. pombe, S. cerevisiae and
human cells, respectively. The biochemical connection
www.sciencedirect.com 0968-0004/$ - see front matter Q 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.tibs.2005.09.005
Review
TRENDS in Biochemical Sciences
Cdk1
297 aa
Cdk2
298 aa
Cdk3
305 aa
Cdk5
292 aa
PCTAIRE3
502 aa
PCTAIRE1
496 aa
PCTAIRE2
523 aa
PFTAIRE1
451 aa
PFTAIRE2
435 aa
Cdk4
303 aa
Cdk6
326 aa
Cdk10
360 aa
PITSLRE
795 aa
CCRK
346 aa
Cdk9
372 aa
CHED
1512 aa
CRK7
1490 aa
Cdk7
346 aa
Cdk8
464 aa
Cdk11
502 aa
Ser/Thr protein kinase
Nuclear localization signal
Vol.30 No.11 November 2005
Coiled-coil domains
631
Ti BS
Figure 1. The mammalian Cdk family. The phylogenetic tree was generated by a bootstrap analysis of the complete amino acid sequences of the human proteins using
CLUSTAL software [91]. The number of amino acids (aa) deduced from the nucleotide sequence and the principal structural domains are indicated for each protein.
between Cdks and cyclins was made in 1989 when
investigators reported the association of Cdk1 with Cyclin
A and Cyclin B in clam oocytes, starfish and X. laevis
oocytes [20]. Additional cyclins in mammals, including
Cyclin C, Cyclin D and Cyclin E, were identified by the
complementation of S. cerevisiae cln mutants or by
cloning chromosomal breakpoints in B-cell lymphomas
(Cyclin D1) [21].
More recently, analysis of the human genome has
identified at least 29 genes encoding related proteins that
share a conserved stretch of 150 amino acid residues
termed the ‘cyclin box’ (Box 1, Figure 2). This domain is
formed by five helical regions and is responsible for
binding to putative partner proteins, including the Cdks
(Figure 2). Although all of these proteins are generically
designated cyclins, for most of them it is not known
whether they are synthesized and destroyed cyclically.
Cdks and cell-cycle progression
In the past decade, the biochemical analysis of a small
group of Cdks, their regulators and their substrates has
provided a general framework for understanding how the
mammalian cell cycle is regulated. After cytokinesis is
completed, the newly generated cells can either continue
cell division or stop proliferating. Cells that choose the
latter option enter into a state that is generally known as
‘quiescence’ or G0, the biochemical parameters of which
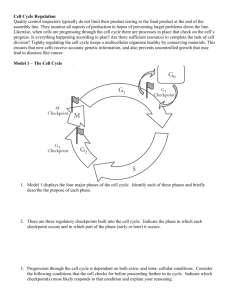
remain poorly defined. Those cells that continue proliferating advance to the G1 phase of the new cycle (Figure 3).
www.sciencedirect.com
G1–S phases
Progression through G1 is regulated by a complex
mechanism that can involve at least three Cdks – Cdk4,
Cdk6 and Cdk2 – and their regulators (Boxes 2,3) [22,23].
Initially, mitogenic signaling induces synthesis of the
D-type cyclins and possibly the proper folding and
transport of Cdk4 and/or Cdk6 to the nucleus. Active
complexes of Cdk4 or Cdk6 and D-type cyclins phosphorylate members of the retinoblastoma (Rb) protein family,
which includes pRb, p107 and p130 (Table 1). These
proteins contain multiple sites for phosphorylation by
Cdks (e.g. pRb has 16), only some of which can be
recognized by the Cdk4–CyclinD and/or Cdk6–CyclinD
complexes. The only other substrates that are known to be
phosphorylated by these complexes are Smad3, Cdt1 and
Marcks, the myristoylated alanine-rich C-kinase substrate (Table 1). So far, in vitro studies have found no
major differences between Cdk4 and Cdk6, or among the
three D-type cyclins. Thus, it is likely that the unique
properties of these molecules are linked to their tissue
specificity and/or their differential activation during
development [24].
Rb proteins function to repress transcription by binding
and modulating the activity of transcription factors, such
as E2F family members, histone deacetylases and
chromatin remodeling complexes [25]. Some of the most
crucial transcriptional targets of Rb–E2F complexes are
the E-type cyclins, which are thought to be required to
activate Cdk2 for proper completion of the G1 phase.
Although the initial phosphorylation of Rb proteins is
PSTAIRE
A1, A2, E1, E2
(D1, D2, B1, B3)
63
42
56
PSTAIRE
PISTVRE
PSSALRE
43
38
PLSTIRE
NRTALRE
E1, E2, A1, A2, C
D1, D2, D3
p35, p39 (D-,Eand G-type
cyclins)
D1, D2, D3
H
28
34
37
26
SMSACRE
PITALRE
PISSLRE
SMSACRE
C (K?)
T1, T2, K
Unknown
L1, L2 (D)
Pctk1, Crk5
35
44
41
45
PITSLRE
PFTAIRE
PFTAIRE
PCTAIRE
Unknown
Unknown
Unknown
Unknown
Pctk2
Pctk3
CrkRS, CD2L7
Cdc2L5
48
45
17
16
PCTAIRE
PCTAIRE
PITAIRE
PITAIRE
(Cables1)
Unknown
Unknown
Unknown
37
PNQALRE
Unknown
Cdc2, Cdc28
CDK2
CDK6
CDK7
CDK8
CDK9
CDK10
CDK11
PITSLRE
PFTAIRE1
PFTAIRE2
PCTAIRE1
PCTAIRE2
PCTAIRE3
CRK7
CHED
CCRK
a
PSK-J3
MO15, CAK,
STK1
K35
Cdc2L1, Cdc2L2
Pftk1
PSTAIRE
Other interacting proteins
Cks
E2F/DP
MyoD
Ets2
14–3-3, 9G8,
CK2, eIF3,
RanBPM,
RNPS1, RNA
pol II
14–3-3, p11,
p35
TRAP
Substrates
Cellular function
Actopaxin, Adenomatous polyposis coli, Amphiphysin1,
APC, BARD1, BRCA2, Caldesmon, Cdc7, Cdc20, Cdc25A,
Cdc25 C, Cdh1, Cdk7, C/EBPb, CKII, Dynein, Dystrophin,
EF-1, Eg5, EGFR, FANCG, Fos, GFAP, GM130, GRASP65,
Histone H1, hHR6A, HMG-I(Y), IFAP300, KRC, Lamins A, B
and C, Lamin B receptor, Lats1, MAP1B, MAP4, Marcks,
MCM2, MCM4, MKLP1, Myb, NBP60, Neurofilament H, NFI, Nir2, Npm, NPC, Nucleolin, Nucks, Numatrin, Orc1, p18,
p47, p53, p54NRB, PAP, Plectin, PP1-I2, pRb, R2, Rab4,
Rap1GAP, RCC1, RIIa, S6K1, Sam68, Separase, Ski,
Survivin, mSTI1, Tau, vimentin, thymidine kinase
BARD1, B-Myb, BRCA1, BRCA2, CBP/p300, Cdc6, Cdc7,
Cdk7, Cdt1, C/EBPb, DP1, hHR6A, HIRA, Ku70, Marcks,
MCM2, MCM4, MyoD, NPAT, Npm, p107, p21Cip1, p27Kip1,
p53, pRb, R2, RPA, Smad3, thymidine kinase
Cables1
Cdt1, Marcks, p107, p130, pRb, Smad3
Amphiphysin1, Cables, Disabled1, Doublecortin, Munc18,
Nudel, p53, Pctaire1, Protein phosphatase inhibitor 1, PSD95, Stat3, mSds3, SynapsinI, tyrosine hydroxylase
p107, p130, pRb
Cdk1–Cdk6, p53, RARg, RNA pol II
Cell cycle (G2–M)
RNA pol II
pRb, RNA pol II
Unknown
9G8, Cyclin L
Unknown
Unknown
Unknown
Unknown
Unknown
Unknown
RNA pol II
Unknown
Unknown
Cell cycle (G1–S)
Cell cycle (G0–G1–S)
Cell cycle (G1–S)
Senescence, postmitotic neurons
Cell cycle (G1–S)
Cdk-activating kinase, transcription
Transcription
Transcription
Transcription, cell cycle (G2–M)
Transcription, Cell cycle (M)
Possible role in apoptosis
Testis- and brain-specific
Unknown
Cell cycle (S–G2), neurite outgrowth
Neuron-specific
Brain-specific
Transcription
Cholinergic signaling and
hematopoietic cell proliferation
Unknown
domain 1; BRCA1, breast cancer 1; CAK, Cdk-activating kinase; CBP/p300; CREB-binding protein/p300; Cdc, cell division cycle; C/EBPb, CCAAT/enhancer-binding protein b; Cks, Cdc28-dependent kinase subunit; CKII, casein kinase II;
DP1, E2F dimerization partner 1; EF-1, elongation factor 1; EGFR, epidermal growth factor receptor; FANCG, Fanconi anemia, complementation group G; GAP, GTPase-activating protein; GFAP, glial fibrillary acidic protein; GM130,
Golgi matrix protein 130; GRASP65, Golgi reassembly stacking protein 1, 65kDa; hHR6A, human homolog of Rad6; HIRA, HIR histone cell-cycle regulation defective homolog A; HMG, high mobility group; IFAP300, intermediate
filament-associated protein, 300 kDa; KRC, kB-binding and recognition component; MAP, microtubule-associated protein; Marcks, myristoylated alanine-rich C-kinase substrate; MCM, minichromosome maintenance; MKLP1,
mitotic kinase-like protein 1; mSTI1, murine stress-inducible protein 1; NBP60, NLS-binding protein 60 kDa; NF-I, nuclear factor I; NPAT, nuclear protein, ataxia-telangiectasia; Npm, nucleophosmin (B23, NO38); NPC, nuclear pore
complex; PAP, phosphatidic acid phosphatase; PP, protein phosphates; pRb, retinoblastoma protein; R2, ribonucleotide reductase 2; RanBPM, Ran-binding protein M; RNPS1, RNA-binding protein S1; RARg, retinoic acid receptor-g;
RCC1, regulator of chromosome condensation 1; RIIa, type II cAMP-dependent protein kinase; RNA pol II, RNA polymerase II; S6K1, S6 kinase; TRAP, TNF receptor-associated protein.Abbreviations: APC: anaphase-promoting
complex; BARD1, BRCA1-associated RING
Vol.30 No.11 November 2005
65
CDK1
Main activating
cyclin (other
cyclins)
A1, A2, B1, B2
(E, B3)
TRENDS in Biochemical Sciences
Cyclin-binding
domain
Synonym
Review
Identity
to Cdk1
(%)
100
Symbol
CDK3
CDK4
CDK5
632
www.sciencedirect.com
Table 1. The mammalian Cdk familya
Review
TRENDS in Biochemical Sciences
Box 1. Cyclins in search of Cdk partners
Of the 29 proteins formally designated as cyclins because they
contain a ‘cyclin box’ (see text), many lack known Cdk (or other
kinase) partners.
† Cyclin F shares the greatest amino acid sequence similarity with
Cyclin A, and its expression fluctuates during the cell cycle with a
pattern similar to those of Cyclin A and Cyclin B [76]. In addition,
Cyclin F has been reported to interact with Cyclin B1 to form trimeric
Cdk1–CyclinB1–CyclinF active complexes [77]. Cyclin F complements
Cdc4 (a component of the SCF proteolytic complex) mutants in yeast
and, similar to Cdc4, contains an F-box domain. Moreover, mice
lacking Cyclin F have indicated that this cyclin might have a potential
role in cell-cycle re-entry from quiescence [78].
† G-type cyclins (G1 and G2) are targets of p53 and seem to be
involved in the ATM–p53–Mdm2 pathway [79]. Although the
expression of Cyclin G1 peaks in G1 and that of Cyclin G2 is highest
in late S phase, these cyclins seem to participate in G2–M arrest in
response to DNA damage [80]. G-type cyclins bind to GAK, a
serine/threonine CyclinG-associated kinase that is involved in
epidermal growth factor receptor signaling [81].
† Cyclin I is most related to the G-type cyclins (Figure 2). The
expression of Cyclin I does not correlate with any of the phases of the
cell cycle; thus, this cyclin might have a that is role unrelated to cellcycle control [82].
† In Drosophila, Cyclin J binds and activates Cdk2 kinase.
Inhibition of this interaction by specific peptide aptamers results in
defects in chromosome segregation and progression through
mitosis [83]. Unfortunately, the corresponding mammalian protein
has not been characterized.
† Other ‘Cdk-orphan’ cyclins, such as the M-type cyclins (M1, M2,
M3, M4) and Cyclin O (also known as Ung2), seem to be involved in
ion transport and DNA repair, respectively [84,85].
† More recently, Cyclin S, a new putative cyclin with structural
similarity to the L-type cyclins, has been described. Cyclin S seems to
be involved in transcriptional changes related to the duration of
memory in neurons [86].
† Finally, analysis of the human genome sequence has identified
seven additional loci that encode putative cyclins on the basis of
sequence homology to cyclin boxes (data not shown). The functions
of the encoded proteins remain to be elucidated.
mediated by the Cdk4–CyclinD and/or Cdk6–CyclinD
kinases, the irreversible inactivation of these proteins is
presumed to be carried out by Cdk2–CyclinE. This process
is believed to render cells independent of mitogenic signals
and corresponds to the ‘restriction point’ [23] (Figure 3).
The ‘restriction point’ has been defined as the stage during
G1 in which cells no longer require mitogenic stimuli to
undergo cell division. Indeed, cells lacking pRb, p107 and
p130 do not have a functional restriction point because
they can divide in the absence of mitogens [26,27].
Another kinase, Cdk3, might also participate in
inactivation of pRb. Cdk3 is highly related to Cdk2 and
Cdk1 (Figure 1) and can complement cdc28 mutants of S.
cerevisiae [9]. Cdk3 interacts with E-type and A-type
cyclins, and also with the Cdk5 and Abl1 enzyme
substrate (Cables; also known as Ik3) subfamily (Figure 2).
So far, however, there is no evidence that Cables proteins
induce the kinase activity of Cdk3; thus, the physiological
significance of this interaction remains to be resolved. It
has been recently shown that Cdk3 also binds Cyclin C
during G0 exit and stimulates phosphorylation of pRb
during the G0–G1 transition of some human tumor cells
[28]. Because the expression of Cyclin C precedes that of
D-type cyclins, inactivation of Rb proteins during early
G1 has been proposed to involve their sequential
www.sciencedirect.com
Vol.30 No.11 November 2005
633
phosphorylation by Cdk3–CyclinC and then Cdk4–
CyclinD and/or Cdk6–CyclinD [28].
Cdk3 must also have Rb-independent roles because a
dominant-negative mutant arrests the cell cycle in the
presence of the SV40 T antigen, a protein that is known to
inactivate Rb proteins [29]. In laboratory mice, the gene
encoding Cdk3 carries a mutation that introduces a
premature stop codon at position 187, and the predicted
truncated protein lacks at least a third of the functional
kinase domain and is presumed to be inactive [30]. Thus,
further studies are needed to understand the precise role
of Cdk3 in the cell cycle.
In addition to phosphorylating Rb proteins, Cdk2–
CyclinE kinase activity is thought to be essential for
initiating DNA replication by facilitating loading of the
MCM chromosome maintenance proteins onto origins of
replication. Once cells enter S phase, Cdk2–CyclinE
complexes need to be silenced to avoid the re-replication
of DNA [31]. This requirement is presumably accomplished, at least in part, by the rapid degradation of Cyclin
E by the SCF–Fbxw7 ubiquitin ligase (Fbxw7 is a F-box
protein also known as Cdc4 or Archipielago), followed by
its subsequent cleavage by the proteasome. Other
substrates known to be phosphorylated by Cdk2–CyclinE
kinases include proteins involved in histone modification
[NPAT (also known as p220), HIRA, CBP/p300], DNA
replication (Cdt1), DNA repair (BRCA1, Ku70) and
centrosome duplication and maturation [CP110, Mps1,
nucleophosmin (also known as B23)]. In addition, Cdk2–
CyclinE phosphorylates its own inhibitor p27Kip1, thereby
facilitating the degradation of this inhibitor by the
proteasome [31].
Inactivation of pRb also participates in promoting the
transcription of genes that are necessary for subsequent
phases of the cell cycle, including those encoding the
A-type and B-type cyclins. Although A-type cyclins
accumulate during S phase, synthesis of B-type cyclins
is not evident until the G2–M transition. Indeed, current
models propose that once Cdk2 is no longer associated
with Cyclin E, it interacts with the newly synthesized
A-type cyclins, A1 and A2 (Figure 3). Whereas Cyclin A2
seems to be ubiquitous, Cyclin A1 is expressed primarily
in germ cells.
Cdk2–CyclinA complexes have been reported to
phosphorylate numerous proteins (Table 1) that are
thought to be required for proper completion and exit
from S phase. These proteins include upstream regulators of Cyclin A (pRb), transcription factors (E2F1,
B-Myb), proteins involved in DNA replication [Cdc6,
HSSB (also known as RPA), MCM4], DNA repair
(BRCA1, Ku70), histone modification (HIRA), ubiquitinmediated proteolysis [hHR6A (also known as Rad6 or
Ubc2) and Cdc20] and cell-cycle checkpoints (p53,
p21Cip1, MDM2) [22]. Whether these phosphorylations
are required for proper cell-cycle progression remains to
be demonstrated (see later).
G2–M phases
At the end of the S phase, A-type cyclins associate with
Cdk1. Cdk2–CyclinA and Cdk1–CyclinA complexes
share several substrates such as proteins involved in
634
Review
TRENDS in Biochemical Sciences
Cyclin F
Cyclin O
Cyclin A1
Cyclin A2
Cyclin B3
Cyclin B1
Cyclin B2
Cyclin D3
Cyclin D1
Cyclin D2
Cyclin E1
Cyclin E2
Cyclin J
Cyclin C
Cyclin H
Cyclin K
Cyclin T1
Cyclin T2
Cyclin L1
Cyclin L2
Cyclin M3
Cyclin M1
Cyclin M2
Cyclin M4
Cables 1
Cables 2
Cyclin I
Cyclin G1
Cyclin G2
N-terminal cyclin box
Vol.30 No.11 November 2005
786 aa
316 aa
465 aa
432 aa
1395 aa
433 aa
398 aa
292 aa
295 aa
289 aa
410 aa
404 aa
372 aa
302 aa
323 aa
355 aa
726 aa
730 aa
526 aa
424 aa
707 aa
586 aa
854 aa
727 aa
633 aa
478 aa
377 aa
295 aa
344 aa
C-terminal cyclin box
F-box
Nuclear localization signal
Proline-rich
Ti BS
Figure 2. Mammalian proteins with a cyclin-box domain. The phylogenetic tree was generated by a bootstrap analysis of the complete amino acid sequences of the human
proteins using CLUSTAL software [91]. The number of amino acids (aa) deduced from the nucleotide sequence and the principal structural domains are indicated for each
protein.
DNA replication (i.e. MCMs, Cdc7 or the ribonucleotide
reductase R2) as well as other proteins that participate
in the control of cell-cycle progression (e.g. pRb, p53,
BARD1 and BRCA2) (Table 1). Whether Cdk2–CyclinA
and Cdk1–CyclinA complexes have differential roles
during the S to G2 transition remains obscure. During
G2, A-type cyclins are degraded by ubiquitin-mediated
proteolysis whereas the B-type cyclins are actively
synthesized. As a consequence, Cdk1 binds to B-type
cyclins – an association believed to be essential for
triggering mitosis. Cdk1 becomes associated with the
B-type cyclins. Cdk1 preferentially binds to the two
main B-type cyclin isoforms, B1 and B2 (Box 2). The role
of Cyclin B3 is not well understood. Although some
reports indicate that its main partner is Cdk1, it has
been suggested that it might function on binding to
Cdk2 [32]. Moreover, it has been recently proposed that
Cyclin B3 might have a specific role in the meiotic cell
cycle [32]. Cdk1–CyclinB complexes are thought to
regulate several events during both the G2–M transition
[33] and progression through mitosis. In fact, these
complexes can phosphorylate more than 70 proteins in
mammalian cells (Table 1). Moreover, the list of putative
Cdk1–CyclinB substrates might be much larger, as
suggested by a recent screening for Cdk1 substrates in
www.sciencedirect.com
yeast [34]. Cytoplasmic Cdk1–CyclinB complexes associate with centrosomes during prophase, where they
promote centrosome separation by phosphorylating the
centrosome-associated motor protein Eg5.
Cdk1–CyclinB complexes are also involved in different regulatory and structural processes, such as
chromosomal condensation (by phosphorylating histones, HMG-I and RIIa), fragmentation of the Golgi
network (by phosphorylating Nir2, p47, GM130 and
GRASP65) and breakdown of the nuclear lamina (by
phosphorylating different lamins, the lamin B receptor
and the nuclear pore complex). Other substrates
(Table 1) include microtubule-binding proteins (dynein,
MAP4, MAP1B, adenomatous polyposis coli, tau),
proteins implicated in replication (MCM2, MCM4,
ribonucleotide reductase R2), translation (S6 kinase,
EF-1) ubiquitin-dependent proteolysis (Cdc20, Cdh1)
and other regulatory proteins involved in mitotic
progression and the exit from mitosis (Cdc25 phosphatases, actopaxin, survivin, Lats1). Finally, the inactivation of Cdk1–CyclinB complexes is required for proper
exit from mitosis. This inactivation is accomplished by
the degradation of B-type cyclins via ubiquitination by
the proteolytic pathway regulated by the anaphasepromoting complex [35].
Review
TRENDS in Biochemical Sciences
CycF
Vol.30 No.11 November 2005
635
Cdk3
Cdk10
CycC
Cdk4
Cdk11
CycD
CycL
G0
Cdk6
CycD
M
Cdk1
G1
CycB
R
G2
S
Cdk2
CycG
CycE
Cdk1
CycA
Cdk2
CycA
Cdk7
CycH
Mat1
CAK
Cdk8
CycC
Ti BS
Figure 3. Proposed roles of Cdk–cyclin complexes in the mammalian cell cycle. Cdk4–CyclinD, Cdk6–CyclinD and Cdk3–CyclinC (at least in human cells) complexes regulate
the G0–G1 transition (in quiescent cells) and the early phases of G1 (in proliferating cells) by phosphorylating the retinoblastoma protein (pRb). Cdk2–CyclinE complexes have
been proposed to complete phosphorylation of pRb, an event that is thought to convey mitogenic independence (passage through the restriction point, R) to dividing cells.
Cdk2–CyclinE complexes have been also implicated in the G1–S transition by licensing DNA origins of replication. Cdk2 later associates with Cyclin A during progression
through S phase. Cdk1 participates in the S–G2 and G2–M transitions by sequential binding to Cyclin A and Cyclin B. These widely accepted roles for the Cdks are indicated by
open arrows. Cdk-activating kinase (CAK) phosphorylates, and presumably activates, all cell-cycle Cdks. CAK, a protein complex formed of Cdk7, CyclinH and Mat1, is a
substrate for Cdk8–CyclinC (filled arrows). Cdk10 and Cdk11 might be involved in mitosis, but their functional relevance is not well understood. Finally, Cyclin F might be
required for entry into G1 and Cyclin G is implicated in the DNA damage response during the G2–M transition (see text). The functions of Cdk3, Cdk10, Cdk11, Cyclin F and
Cyclin G are represented by dotted arrows to indicate that the data that implicate them in the cell cycle are still preliminary.
Biological role of other Cdks
Cdk5 is activated by p35 and p39, two proteins that are
almost uniquely expressed in brain [36,37]. Cdk5 also
binds to D-type and E-type cyclins although the heterodimeric complexes do not have kinase activity [36]. Cdk5–
p35 and Cdk5–p39 complexes phosphorylate numerous
substrates (Table 1) involved in several aspects of
transcription (mSds3, Stat3, p53), neuronal function
(tyrosine hydroxylase, Nudel, Amphiphysin 1, Munc18a),
migration (Doublecortin, Disabled1) and synaptic transmission (Synapsin1, PSD-95) [37].
Cdk7 to Cdk11 show activities related to the control of
transcription, in some cases with direct implications in
cell-cycle control. Cdk7 is a component of the Cdkactivating kinase (CAK), which phosphorylates and
presumably activates all cell-cycle Cdks (Table 1 and
Box 3). Moreover, CAK, along with six additional subunits,
forms the general transcription factor TFIIH that is
www.sciencedirect.com
involved in promoter clearance and progression of
transcription [38]. Cdk8–CyclinC and Cdk9–CyclinT
complexes also regulate transcription by phosphorylating
the C-terminal domain of the large subunit of RNA
polymerase II. Indeed, Cdk8–CyclinC complexes are
components of the RNA polymerase holoenzyme [39].
Cdk8–CyclinC also phosphorylates Cyclin H to inhibit
CAK activity. Cdk9, by contrast, binds to Cyclin T and
Cyclin K to form the P-TEFb transcription factors
implicated in transcript elongation by RNA polymerase
II [40].
Cdk10 is thought to be involved in regulating the G2–M
phase of the cell cycle because Cdk10 antisense and
dominant-negative mutants arrest cells in G2–M; however, no cyclin partner for this kinase has been identified
so far (Box 2). Cdk10 also inhibits transactivation of the
Ets2 transcription factor, a regulator of Cdk1 expression
[41]. Finally, Cdk11 binds to Cyclin L and interacts with
636
Review
TRENDS in Biochemical Sciences
Vol.30 No.11 November 2005
Box 2. Summary of Cdk–Cyclin complexes
Box 3. Regulation of cell-cycle Cdks
Cdk1 binds preferentially to A-type (A1 and A2) and B-type (B1, B2
and B3) cyclins [22,23,87]. In addition, Cdk1 might form trimeric
complexes with Cyclin B and Cyclin F [77]. Cyclin B3, albeit most
homologous to Cyclin B1 and Cyclin B2, has some properties that
resemble those of A-type cyclins, including association with Cdk2
[32]. Cdk1 also forms active complexes with E-type cyclins, at least in
cells deficient for both Cdk2 and p27Kip1 [52]. The main partner of the
E-type cyclins is Cdk2, which also recognizes the A-type cyclins. In
addition, Cdk2 can bind to D-type cyclins [45], and to Cyclin B1 [52].
Cdk3 binds to E-type and A-type cyclins, which suggests that it has
a role similar to that of Cdk2. In addition, Cdk3, but not Cdk2, binds to
Cyclin C during the G0–G1 transition [28]. The only known partners
for Cdk4 and Cdk6 are the D-type cyclins (D1–D3). Cdk5 has two
activating partners, p35 and p39, none of which is a cyclin [36,37]. In
addition, Cdk5 has been reported to bind to D-type and G-type
cyclins. Notably, Cyclin G also recognizes a non-Cdk kinase
designated ‘G-cyclin-associated kinase’ (GAK) [81].
Cdk7 is a subunit, along with Cyclin H and Mat1, of the Cdkactivating kinase (CAK) [38]. The only known partner of Cdk8 is
Cyclin C [39]. Cdk9 binds to Cyclin K and the T-type cyclins (T1, T2)
[40], whereas Cdk11 recognizes the L-type cyclins (L1, L2) [42].
Finally, Cdk10 binds to the transcription factor Ets2 [41], but there are
no known cyclin partners for this catalytic subunit. Likewise, various
cyclins, including Cyclin I, Cyclin J, M-type cyclins (M1–M4), Cyclin
O, Cyclin P and Cyclin S, do not have known Cdk partners.
The kinase activity of cell-cycle Cdks is tightly controlled at different
levels, including interaction with activating subunits (cyclins),
binding to negative regulators (Cdk inhibitors or CKIs), phosphorylation–dephosphorylation, folding and subcellular localization [22,23].
Two families of Cdk inhibitors have been described: the INK4
family (p16INK4a, p15INK4b, p18INK4c, p19INK4d), and the Cip and Kip
family (p21Cip1, p27Kip1, p57Kip2) [22]. INK4 proteins specifically bind
to and inhibit monomeric Cdk4 and Cdk6 proteins (Figure I). Cip and
Kip proteins, by contrast, bind to Cdk–cyclin complexes [22,88].
Binding to Cdk2 and Cdk1 complexes blocks the kinase activity of
these complexes; however, the role of Cip or Kip binding to Cdk4–
CyclinD or Cdk6–CyclinD complexes is unclear. Early observations
indicating that p21Cip1and p27Kip1 are essential for the formation of
these complexes [22] have not been confirmed [89,90]. Moreover,
these inhibitors can block the kinase activity of Cdk4–CyclinD and
Cdk6–CyclinD, but not at stochiometric concentrations [22]. Recent
genetic evidence indicates that Cip and Kip inhibitors can block the
cell cycle in the absence of either Cdk2 or Cdk4 and Cdk6 [45,51];
however, compensatory activities between these Cdks and from
Cdk1 have hindered the unequivocal identification of the physiological targets of Cip and Kip inhibitors.
Cdk kinase activity is also controlled by activating and/or inhibitory
phosphorylations and dephosphorylations [23]. Whereas the Wee1 and
Myt1 kinases inhibit the kinase activity of Cdk–cyclin complexes by
phosphorylating adjacent threonine and tyrosine residues in the Cdk
subunit, Cdc25 phosphatases (Cdc25A, Cdc25B and Cdc25C) activate
these kinases by dephosphorylating the very same amino acid residues.
In addition, active Cdk–cyclin complexes need to be phosphorylated in
the T-loop of the Cdk subunit by Cdk-activating kinase. If both activating
and inactivating phosphorylations exist in the same molecule, they
result in an inactive kinase. Additional levels of regulation, such as
protein folding and subcellular localization, are not considered here.
the general precursor mRNA splicing factors RNPS1 and
9G8. Cdk11–CyclinL also interacts with RNA polymerase
II, playing a role in transcript production and regulation of
RNA processing [42]. It is likely that this group of Cdks
link growth factor signaling pathways to transcription
and RNA processing events in a manner dependent on the
cell cycle [40,42].
Information regarding the role of the other nine members
of the Cdk structural family, for which no partners have been
identified, is still limited. The most relevant information
relating to these kinases is summarized in Table 1.
Genetic analysis of the role of Cdks in mice
The role of Cdks outlined above has been primarily
deduced from biochemical studies that have mainly used
human tumor cell lines. More recently, the role of these
kinases has been investigated by genetic approaches using
gene-targeting strategies in mice (Table 2). Below, we
summarize these findings and discuss their implications
with a particular emphasis on our understanding of how
Cdks control cell-cycle progression.
Inactive
P
Cdk
Cyc
Cyc
Wee1
Myt1
Cdc25
Inactive
Cdk
Cdk
Cyc
Cdk7
CycH
INK4
Mat1
CKIs
Cdk4–CyclinD and Cdk6–CyclinD complexes
Ablation of either Cdk4 or Cdk6 is compatible with life, at
least in mice [43–45]. Indeed, inactivation of the genes
encoding these kinases affects the proliferation of only
specific types of cell (Table 2). For example, loss of Cdk4
expression prevents the postnatal proliferation of pancreatic b cells and pituitary lactotrophs, but it has no effect
on the neogenesis of these types of cell from embryonic
precursors [43,44,46,47]. Likewise, the ablation of Cdk6
results in limited defects in the hematopoietic compartment [45]. In the main, these Cdk6-deficient mice show a
slight decrease in peripheral and spleen red blood cells.
The limited requirements for these Cdks could be due to
compensatory mechanisms; however, the ablation of both
www.sciencedirect.com
Cdk
Cip/Kip
CAK
P
Cyc
Active
Ti BS
Figure I. Basic regulatory mechanisms of cell-cycle Cdks. These Cdks require
binding to their cyclin partners for activation of their kinase activity. Some Cdks
(Cdk4 and Cdk6) are inhibited by direct binding of the INK4 family of CKIs. By
contrast, Cip and Kip inhibitors block kinase activity by forming inactive trimeric
complexes (Cdk2–CyclinE, Cdk2–CyclinA, Cdk1–CyclinA, Cdk1–CyclinB, and
possibly Cdk4–CyclinD and Cdk6–CyclinD). Cdk–cyclin complexes can be activated
by phosphorylation in their conserved T-loop of t he Cdk subunit by CAK. By
contrast, Cdk–Cyclin complexes can be negatively regulated by phosphorylation
in adjacent threonine or tyrosine residues by the dual-specificity kinases Wee1 and
Myt1. These inhibitory phosphorylations can be reversed by the dual-specificity
Cdc25 phosphatases that act as positive regulators of Cdk–cyclin activity.
Review
TRENDS in Biochemical Sciences
Vol.30 No.11 November 2005
637
Table 2. Mouse models of mammalian Cdksa
Cdk
Cdk1
Mutation
Null
Viability
Lethal
Phenotype in vivo
N/A
Phenotype in vitro
N/A
Cdk2
Null
Viable
Cdk2
Viable
Early senescence in MEFs in culture;
no major cell-cycle defects; does not
rescue proliferative alterations by
p21Cip1 or p27Kip1 loss
No defects in cell proliferation after
ablation
Normal; mutation present in ‘wild
type’ MEFs
Faster cell cycles and no ‘culture crisis’;
increased susceptibility to transformation by ras oncogenes
Cdk4
Conditional
null
Premature
stop codon
R42C mutant
insensitive
to INK4
inhibitors
Null
Male and female sterility due to meiotic defects; no defects in mitotic cells;
does not rescue organomegalia or
tumors induced by p27Kip1 loss
No phenotype observed
Cdk5
Null
Cdk6
Null
Perinatal
lethality
Viable
Cdk2 Cdk6
Double null
Viable
Cdk4 Cdk6
Double null
Late
embryonic
lethality
Cdk11
Null
Lethal E3.5
Cdk3
Cdk4
a
Viable
Viable
Viable
Normal; most laboratory strains carry
this mutation
Epithelial and mesenchymal tumors
with complete penetrance after 14–18
months
Lack of proliferation of postnatal pancreatic b cells and pituitary lactotrophs;
small size
Defective development and structure
of the nervous system
Defective erythroid lineage development
Phenotype identical to Cdk2 and Cdk6
single mutants
Limited proliferation of committed
hematopoietic precursors, especially
those of erythroid origin
Mitotic defects; dead at blastocyst
stage
Refs
M. Malumbres
M. Barbacid,
unpublished
observations
[49–52]
[49]
[30]
[43,69,70]
Decreased susceptibility to immortalization or transformation by oncogenes
[43,44,74]
N/A
[58–61]
No phenotype in MEFs but delayed
proliferation of lymphoid cells
Phenotype identical to Cdk2 and Cdk6
single mutants; no synergism
Delayed cell cycles and decreased pRb
phosphorylation; cells respond to
mitogenic stimuli and became immortal on passage
Proliferative defects and apoptosis in
embryos in culture
[45]
[45]
[45]
[64]
Abbreviations: E, embryonic day; MEF, mouse embryonic fibroblast; N/A, not analyzed; pRb, retinoblastoma protein.
Cdk4 and Cdk6 in mouse embryos has little effect on
overall cell proliferation and organogenesis. Embryos that
are doubly deficient for Cdk4 and Cdk6 develop past
midgestation, although they are not viable at later stages
[embryonic day 18.5 (E18.5)] owing to a proliferative
defect in their erythroid lineage that leads to severe
anemia [45]. Moreover, these double mutant embryos have
limited numbers of early progenitors of both lymphoid and
myeloid lineages in the spleen. These results suggest that
Cdk4 compensates partially for the absence of Cdk6 in
hematopoietic cells. These Cdks, however, are not
required for cell proliferation and/or the differentiation
of most cell types, at least during embryonic development.
The generation of conditional Cdk4 mutant mice should
make it possible to determine whether these kinases are
also dispensable for cell proliferation during adult
homeostasis.
Interestingly, ablation of the three D-type cyclins leads
to an almost identical phenotype although the mice die
slightly earlier [48]. The longer survival of the double
Cdk4 Cdk6 mutant embryos might be due to the formation
of Cdk2–CyclinD complexes [45], although other reasons,
such as differences in genetic background, cannot be ruled
out at present.
The absence of Cdk4 and Cdk6 (or the three D-type
cyclins) does not prevent the proliferation of mouse
embryonic fibroblast (MEF) in culture; however, these
double mutant cells show decreased proliferation rates,
accompanied by diminished phosphorylation of pRb and
delayed expression of S- and M-phase markers [45,48].
Interestingly, Cdk4;Cdk6 double mutant MEFs respond to
www.sciencedirect.com
mitogenic stimuli after serum starvation, indicating that
Cdk4–CyclinD and Cdk6–CyclinD complexes contribute
to, but are not essential for mitogen-induced proliferation
or to exit from quiescence. These findings underscore the
need to search for additional pathways implicated in the
proliferative response of cells to mitogenic stimuli. Finally,
Cdk4;Cdk6 double null cells (as well as those lacking
D-type cyclins) respond to cell-cycle inhibition by Cip or
Kip inhibitors, but are insensitive to INK4 proteins
[45,48]. These observations confirm that INK4 inhibitors
are specific for Cdk4–CyclinD and Cdk6–CyclinD
complexes.
Cdk2
On the one hand, genetic studies have established that
Cdk2 is not required for the mitotic cell cycle because mice
lacking this enzyme survive for over 2 years without
obvious defects [49,50]. These observations challenge the
central role in key cell-cycle events that has been
attributed to this Cdk. On the other hand, Cdk2 is
essential for the survival of germ cells – a previously
unsuspected activity [49]. In the absence of Cdk2, male
germ cells die in the first meiotic division owing to defects
in chromosome pairing that presumably activate the
pachytene checkpoint. Oocytes also die in the first meiotic
division, albeit at the dyctyate stage [49]. The targets of
Cdk2 that are essential for meiotic cell division remain to
be identified.
The role of Cdk2 in mediating the cell-cycle inhibitory
and tumor-suppressing activities of p21Cip1 and p27Kip1 is
also dispensable [51,52]. For example, p21Cip1 and p27Kip1
638
Review
TRENDS in Biochemical Sciences
induce cell-cycle arrest regardless of whether cells express
Cdk2 or not. Moreover, the combined deficiency of Cdk2
with either protein results in increased proliferation rates
similar to those of p27Kip1-null or p21Cip1-null cells [51].
Likewise, Cdk2;p21Cip1 double mutant cells fail to arrest
in G1 after DNA damage [51]. More notably, Cdk2;p27Kip1
double mutant mice develop pituitary tumors with the
same high penetrance and latency as those formed in
p27Kip1 single mutant mice [51,52].
The dispensability of Cdk2 might be explained by
compensatory activities of other kinases, most likely the
activity of Cdk1 through its association with Cyclin A, a
partner of Cdk2. Moreover, recent findings indicate that
Cdk1 might also associate with E-type cyclins, at least in
the absence of Cdk2 and p27Kip1 [52]. These hypotheses do
not explain, however, why Cdk1 (or other Cdks) can
compensate so readily for the absence of Cdk2 in the
mitotic cell cycle, but not in meiosis. Alternatively, it is
possible that Cdk2 evolved primarily as a meiotic kinase
with a secondary role in the mitotic cell cycle. Genetic
studies have failed to find compensatory activity from
Cdk6 and Cdk3 because Cdk2;Cdk6 double mutant mice
show the same defects as their parental strains [45] and
Cdk3 is already mutated in Cdk2-null mice [30,49].
Similar studies with Cdk4 have been hampered by the
close genetic localization of the cdk2 and cdk4 loci on
chromosome 10. Finally, crosses between Cdk2-null mice
and mice deficient in other Cdks have not been reported.
Loss of both E-type cyclins results in embryonic
lethality because of defective trophoblast endoreplication
[53,54]. In vitro, cells without E-type cyclins proliferate
well, although they fail to exit quiescence owing to a defect
in the loading of MCM proteins onto DNA pre-replication
complexes [54]. These findings indicate that E-type cyclins
have functions that are independent of Cdk2. Whether
Cdk1 mediates these functions, as has been recently
suggested [52], awaits genetic scrutiny.
Cdk1
So far there are no reports of Cdk1 mutant mice.
Preliminary observations indicate, however, that the
ablation of Cdk1 might result in embryonic lethality (M.
Malumbres and M. Barbacid, unpublished observations).
Similarly, the ablation of its partners Cyclin A2 [55] and
Cyclin B1 [56] also leads to embryonic lethality. By
contrast, the deletion of Cyclin A1 results in male sterility
[57], suggesting that this cyclin has a specific role in germ
cells. Mice lacking Cyclin B2 do not show significant
defects [56].
Other Cdks
Targeted disruption of the cdk5 locus in mice results in
embryonic lethality, although a few pups make it to term
but die a few hours later [58,59]. Cdk5-null embryos show
massive abnormalities in the development and structure
of their nervous system. Re-expression of Cdk5 under the
neuronal-specific p35 promoter reverses all of the defects
[60]. Moreover, p35;p39 double mutant mice show a
similar phenotype to that of Cdk5-defective mice,
suggesting that Cdk5 kinase activity requires these
brain-specific subunits [61].
www.sciencedirect.com
Vol.30 No.11 November 2005
Neither Cdk7 nor its partner, Cyclin H, has been
knocked out in mice; however, disruption of Mat1, the
third component of CAK, results in pre-implantation
lethality (at E3.5) that coincides with depletion of the
maternal Mat1 protein [62]. Mat1-deficient blastocysts
fail to proliferate but retain transcriptional and translational integrity [62,63], which suggests that, although it
might have specific functions in transcription, Mat1 does
not seem to be necessary for transcription mediated by
RNA polymerase II. Whether the cell-cycle defect is due to
a failure of the CAK complex to activate one or more cellcycle Cdks remains to be determined.
Loss of Cdk11 also results in embryonic lethality before
implantation [64]. Cdk11-deficient blastocysts show
growth retardation and die as a result of mitotic arrest
and subsequent apoptosis. A comprehensive list of mice
that have mutations in other Cdk regulators is provided in
recent reviews [24,65].
Cdks as targets for cancer therapy
Cdks are seldom mutated in human cancer. Exceptions
include a miscoding mutation in Cdk4 that renders this
kinase resistant to INK4 inhibitors in a small percentage
of familial melanomas [66] and a few cases of splenic
marginal zone lymphoma and B-cell lymphoma in which
Cdk6 is translocated near the immunoglobulin loci [67,68].
Knock-in mice carrying this very same mutation in Cdk4
(the Cdk4R24C strain) develop diverse tumors, including
mesenquimal, endocrine and other epithelial malignancies [69]. Moreover, exposing these mice to the classical
skin carcinogenesis protocol of 7,12-dimethylbenzanthracene plus 12-O-tetradecanoylphorbol-13-acetate (DMBA
plus TPA) results in the development invasive melanoma
with complete penetrance [70] (Table 2).
Most human tumors carry mutations or epigenetic
alterations in upstream regulators of Cdks (mainly INK4,
Cip and Kip inhibitors, and D-type and E-type cyclins) or
in their substrates (mainly pRb) [23]. Thus, Cdks have
been considered as targets for therapeutic intervention for
more than a decade [71]. Indeed, Cdk inhibitors showing a
different range of specificities are currently undergoing
clinical trials. Unfortunately, in our opinion there is still
insufficient information to predict whether blocking Cdks
will provide therapeutic benefit and, if so, what strategy
(e.g. one targeted towards various Cdks, or one targeted
towards, for instance, Cdk4 rather than Cdk2) will provide
the best results.
Genetic evidence suggests that blocking Cdk4–CyclinD
complexes might result in at least some therapeutic
benefit. For example, mice lacking Cyclin D1 are resistant
to mammary tumors induced by the ras and erbB2
oncogenes [72]. In addition, Cdk4 mutant mice show a
lower incidence of tumor development in various experimental systems [73,74]. However, these mice lack Cdk4 in
all tissues and were generated by the ablation of Cdk4 in
the germ line. Thus, these mice might not be a suitable
model in which to predict the therapeutic value of
inhibiting Cdk4–CyclinD complexes. For example, Cyclin
D1 knockout mice might be resistant to ras or erbB2
because they lack the mammary gland stem cells that are
presumably targeted by these oncogenes. Likewise,
Review
TRENDS in Biochemical Sciences
Cdk4-null mice are small and lack certain endocrine cells
[43,44,46,47], a phenotype that could contribute to the
reduced tumor development observed in these mice.
Most drug discovery efforts have been directed against
Cdk2, because this enzyme has been thought to be
essential for the early phases of the cell cycle that are
most often misregulated in tumor cells. Recent genetic
evidence indicating that Cdk2 is not necessary for normal
development [49,50] has opened up an interesting debate.
Some would argue that these observations are good news
because selective Cdk2 inhibitors will not be toxic. Others,
however, would argue that if Cdk2 is not essential for
normal cell division then it is unlikely to be involved in
tumor cell proliferation. The availability of conditional
Cdk2 knockout mice [49] will enable us to determine
experimentally whether the specific ablation of Cdk2 in
tumor tissue provides any therapeutic advantage. We
must not forget, however, that inhibitors render Cdk2
inactive rather than prevent its expression. Notably,
ectopic expression of a dominant-negative mutant of
Cdk2 blocks proliferation in a few human tumor cell
lines [75]; however, the molecular bases for the differential
sensitivity of human tumor cells to these reagents remains
to be determined.
The development of a new generation of conditional
strains of knock-in mice in which Cdks could be rendered
inactive in tumor tissue should provide valuable information to predict whether the selective inhibition of Cdks
will provide any therapeutic benefit, at least in those
tumors showing a misregulated cell cycle. Hopefully, the
systematic combination of biochemical and genetic studies
using more physiological model systems that closely
mimic the human disease should help us to better validate
potential targets and identify those selective inhibitors
that are most likely to produce a therapeutic benefit in
individuals with cancer.
Concluding remarks
The Cdk family of mammalian kinases encompasses 20
proteins of which 10, those encoding Cdk1 to Cdk9 and
Cdk11, have been formally shown to be part of heterodimeric serine/threonine kinases that require a regulatory
subunit for biological activity. Among these, only five –
Cdk1, Cdk2, Cdk3, Cdk4 and Cdk6 – have partners that
can be properly designated ‘cyclins’ because they are
synthesized and degraded in a cyclical fashion every cell
division. Functional characterization of these heterodimeric kinases has identified roles in driving the basic
machinery of the cell cycle (Cdk1, Cdk2, Cdk3, Cdk4,
Cdk6), regulation of the cell cycle (Cdk7), transcription
(Cdk7 to Cdk11) and neuronal differentiation (Cdk5).
Biochemical analysis of the cell-cycle Cdks in the past
15 years has led to a widely accepted model of mammalian
cell division in which specific Cdks are responsible for
driving each of the cell-cycle phases (Figure 3): G1 (Cdk4,
Cdk6), G1–S (Cdk2) and G2–M (Cdk1). Recent genetic
evidence obtained by targeting the loci encoding these
Cdks in murine embryonic stem cells, however, is
challenging some of the basic concepts of this model
as follows:
www.sciencedirect.com
Vol.30 No.11 November 2005
639
(i) although Cdk4, Cdk6 and Cdk2 are not essential for
the basic cell cycle, these enzymes are absolutely
required for the proliferation of certain specialized
cells. Whereas the role of Cdk6 in hematopoietic
cells could be predicted from its pattern of
expression, the essential role of Cdk4 in some
endocrine cells and that of Cdk2 in germ cells
could not be predicted from previous biochemical
and expression studies.
(ii) Cdk4 and Cdk6 are not essential for entry into the
cell cycle on mitogenic stimuli. These findings
should stimulate further research on additional
molecular mechanisms that might regulate exit
from quiescence.
(iii) Cdk2 is not essential for the mitotic cell cycle or for
mediating the inhibitory and tumor suppressor
activities of p21Cip1 and p27Kip1. Indeed, genetic
studies carried so far have failed to demonstrate a
major role for Cdk2 in mitotic cell division.
Future studies need to expand current genetic information to determine to what extent some of these findings,
in particular those of a negative nature, are due to
compensatory activities from other cell-cycle Cdks or
even by other members of the Cdk family. These studies
will require the generation of double or triple mutant
strains and will take some time. Finally, genetic analysis
should be extended to tumors by means of developing
conditional knockout, or even better conditional knock-in,
strains. Such mice will enable investigators to inactivate
these enzymes directly in tumor tissue by genetic means.
These studies should provide crucial information to devise
the best possible strategies to target these kinases for
therapeutic purposes in individuals with cancer.
Acknowledgements
Work in our laboratories is supported by the V European Framework, the
Spanish Ministry of Science, Comunidad de Madrid, Fundación Ramón
Areces, Fundación La Caixa and the Spanish Association Against Cancer
(AECC). The CNIO is supported by the Fondo de Investigaciones
Sanitarias (RTICCC C03/10).
References
1 Russell, P. and Nurse, P. (1986) Schizosaccharomyces pombe and
Saccharomyces cerevisiae: a look at yeasts divided. Cell 45, 781–782
2 Draetta, G. et al. (1987) Identification of p34 and p13, human
homologs of the cell cycle regulators of fission yeast encoded by
cdc2C and suc1C. Cell 50, 319–325
3 Lee, M.G. and Nurse, P. (1987) Complementation used to clone a
human homologue of the fission yeast cell cycle control gene cdc2.
Nature 327, 31–35
4 Hanks, S.K. (1987) Homology probing: identification of cDNA clones
encoding members of the protein-serine kinase family. Proc. Natl.
Acad. Sci. U. S. A. 84, 388–392
5 Elledge, S.J. and Spottswood, M.R. (1991) A new human p34 protein
kinase, CDK2, identified by complementation of a cdc28 mutation in
Saccharomyces cerevisiae, is a homolog of Xenopus Eg1. EMBO J. 10,
2653–2659
6 Paris, J. et al. (1991) Cloning by differential screening of a Xenopus
cDNA coding for a protein highly homologous to cdc2. Proc. Natl.
Acad. Sci. U. S. A. 88, 1039–1043
7 Tsai, L.H. et al. (1991) Isolation of the human cdk2 gene that encodes
the cyclin A- and adenovirus E1A-associated p33 kinase. Nature 353,
174–177
8 Ninomiya-Tsuji, J. et al. (1991) Cloning of a human cDNA encoding a
CDC2-related kinase by complementation of a budding yeast cdc28
mutation. Proc. Natl. Acad. Sci. U. S. A. 88, 9006–9010
640
Review
TRENDS in Biochemical Sciences
9 Meyerson, M. et al. (1992) A family of cdc2-related protein kinases.
EMBO J. 11, 2909–2917
10 Matsushime, H. et al. (1992) Identification and properties of an
atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type
G1 cyclins. Cell 71, 323–334
11 Xiong, Y. et al. (1992) D type cyclins associate with multiple protein
kinases and the DNA replication and repair factor PCNA. Cell 71,
505–514
12 Hellmich, M.R. et al. (1992) Neuronal cdc2-like kinase: a cdc2-related
protein kinase with predominantly neuronal expression. Proc. Natl.
Acad. Sci. U. S. A. 89, 10867–10871
13 Fisher, R.P. and Morgan, D.O. (1994) A novel cyclin associates
with MO15/CDK7 to form the CDK-activating kinase. Cell 78,
713–724
14 Tassan, J.P. et al. (1995) Identification of human cyclin-dependent
kinase 8, a putative protein kinase partner for cyclin C. Proc. Natl.
Acad. Sci. U. S. A. 92, 8871–8875
15 Graña, X. et al. (1994) PITALRE, a nuclear CDC2-related protein
kinase that phosphorylates the retinoblastoma protein in vitro. Proc.
Natl. Acad. Sci. U. S. A. 91, 3834–3838
16 Graña, X. et al. (1994) PISSLRE, a human novel CDC2-related protein
kinase. Oncogene 9, 2097–2103
17 Xiang, J. et al. (1994) Molecular cloning and expression of
alternatively spliced PITSLRE protein kinase isoforms. J. Biol.
Chem. 269, 15786–15794
18 Evans, T. et al. (1983) Cyclin: a protein specified by maternal mRNA in
sea urchin eggs that is destroyed at each cleavage division. Cell 33,
389–396
19 Hunt, T. (1989) Maturation promoting factor, cyclin and the control of
M-phase. Curr. Opin. Cell Biol. 1, 268–274
20 Nurse, P. (1990) Universal control mechanism regulating onset of
M-phase. Nature 344, 503–508
21 Xiong, Y. and Beach, D. (1991) Population explosion in the cyclin
family. Curr. Biol. 1, 362–364
22 Sherr, C.J. and Roberts, J.M. (1999) CDK inhibitors: positive and
negative regulators of G1-phase progression. Genes Dev. 13,
1501–1512
23 Malumbres, M. and Barbacid, M. (2001) To cycle or not to cycle: a
critical decision in cancer. Nat. Rev. Cancer 1, 222–231
24 Ciemerych, M.A. and Sicinski, P. (2005) Cell cycle in mouse
development. Oncogene 24, 2877–2898
25 Cobrinik, D. (2005) Pocket proteins and cell cycle control. Oncogene
24, 2796–2809
26 Dannenberg, J-H. et al. (2000) Ablation of the retinoblastoma gene
family deregulates G1 control causing immortalization and increased
cell turnover under growth-restricting conditions. Genes Dev. 14,
3051–3064
27 Sage, J. et al. (2000) Targeted disruption of the three Rb-related
genes leads to loss of G1 control and immortalization. Genes Dev. 14,
3037–3050
28 Ren, S. and Rollins, B.J. (2004) Cyclin C/Cdk3 promotes Rb-dependent
G0 exit. Cell 117, 239–251
29 Hofmann, F. and Livingston, D.M. (1996) Differential effects of cdk2
and cdk3 on the control of pRb and E2F function during G1 exit. Genes
Dev. 10, 851–861
30 Ye, X. et al. (2001) A premature-termination mutation in the Mus
musculus cyclin-dependent kinase 3 gene. Proc. Natl. Acad. Sci. U. S.
A. 98, 1682–1686
31 Hwang, H.C. and Clurman, B.E. (2005) Cyclin E in normal and
neoplastic cell cycles. Oncogene 24, 2776–2786
32 Nguyen, T.B. et al. (2002) Characterization and expression of
mammalian cyclin B3, a prepachytene meiotic cyclin. J. Biol. Chem.
277, 41960–41969
33 Nigg, E.A. (2001) Mitotic kinases as regulators of cell division and its
checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32
34 Ubersax, J.A. et al. (2003) Targets of the cyclin-dependent kinase
Cdk1. Nature 425, 859–864
35 Harper, J.W. et al. (2002) The anaphase-promoting complex: it’s not
just for mitosis any more. Genes Dev. 16, 2179–2206
36 Kesavapany, S. et al. (2004) Neuronal cyclin-dependent kinase 5: role
in nervous system function and its specific inhibition by the Cdk5
inhibitory peptide. Biochim. Biophys. Acta 1697, 143–153
www.sciencedirect.com
Vol.30 No.11 November 2005
37 Cruz, J.C. and Tsai, L.H. (2004) A Jekyll and Hyde kinase: roles for
Cdk5 in brain development and disease. Curr. Opin. Neurobiol. 14,
390–394
38 Lolli, G. and Johnson, L.N. (2005) CAK – cyclin-dependent activating
kinase: a key kinase in cell cycle control and a target for drugs? Cell
Cycle 4, 572–577
39 Akoulitchev, S. et al. (2000) TFIIH is negatively regulated by cdk8containing mediator complexes. Nature 407, 102–106
40 Garriga, J. and Graña, X. (2004) Cellular control of gene expression by
T-type cyclin/CDK9 complexes. Gene 337, 15–23
41 Kasten, M. and Giordano, A. (2001) Cdk10, a Cdc2-related kinase,
associates with the Ets2 transcription factor and modulates its
transactivation activity. Oncogene 20, 1832–1838
42 Loyer, P. et al. (2005) Role of CDK/cyclin complexes in transcription
and RNA splicing. Cell. Signal. 17, 1033–1051
43 Rane, S.G. et al. (1999) Loss of Cdk4 expression causes insulindeficient diabetes and Cdk4 activation results in b-cell hyperplasia.
Nat. Genet. 22, 44–52
44 Tsutsui, T. et al. (1999) Targeted disruption of Cdk4 delays cell cycle
entry with enhanced p27Kip1 activity. Mol. Cell. Biol. 19, 7011–7019
45 Malumbres, M. et al. (2004) Mammalian cells cycle without the D-type
cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504
46 Martin, J. et al. (2003) Genetic rescue of Cdk4 null mice restores
pancreatic b-cell proliferation but not homeostatic cell number.
Oncogene 22, 5261–5269
47 Moons, D.S. et al. (2002) Pituitary hypoplasia and lactotroph
dysfunction in mice deficient for cyclin-dependent kinase-4. Endocrinology 143, 3001–3008
48 Kozar, K. et al. (2004) Mouse development and cell proliferation in the
absence of D-cyclins. Cell 118, 477–491
49 Ortega, S. et al. (2003) Cyclin-dependent kinase 2 is essential for
meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31
50 Berthet, C. et al. (2003) Cdk2 knockout mice are viable. Curr. Biol. 13,
1775–1785
51 Martin, A. et al. (2005) Cdk2 is dispensable for cell cycle inhibition and
tumor suppression mediated by p27Kip1 and p21Cip1. Cancer Cell 7,
591–598
52 Aleem, E. et al. (2005) Cdc2–cyclin E complexes regulate the G1/S
phase transition. Nat. Cell Biol. 7, 831–836
53 Parisi, T. et al. (2003) Cyclins E1 and E2 are required for
endoreplication in placental trophoblast giant cells. EMBO J. 22,
4794–4803
54 Geng, Y. et al. (2003) Cyclin E ablation in the mouse. Cell 114, 431–443
55 Murphy, M. et al. (1997) Delayed early embryonic lethality following
disruption of the murine cyclin A2 gene. Nat. Genet. 15, 83–86;
erratum ibid (1999) Nat. Genet. 23, 481
56 Brandeis, M. et al. (1998) Cyclin B2-null mice develop normally and
are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad.
Sci. U. S. A. 95, 4344–4349
57 Liu, D. et al. (1998) Cyclin A1 is required for meiosis in the male
mouse. Nat. Genet. 20, 377–380
58 Ohshima, T. et al. (1996) Targeted disruption of the cyclin-dependent
kinase 5 gene results in abnormal corticogenesis, neuronal pathology
and perinatal death. Proc. Natl. Acad. Sci. U. S. A. 93, 11173–11178
59 Nikolic, M. et al. (1996) The cdk5/p35 kinase is essential for neurite
outgrowth during neuronal differentiation. Genes Dev. 10, 816–825
60 Tanaka, T. et al. (2001) Neuronal cyclin-dependent kinase 5 activity is
critical for survival. J. Neurosci. 21, 550–558
61 Ko, J. et al. (2001) p35 and p39 are essential for cyclin-dependent
kinase 5 function during neurodevelopment. J. Neurosci. 21,
6758–6771
62 Rossi, D.J. et al. (2001) Inability to enter S phase and defective RNA
polymerase II CTD phosphorylation in mice lacking Mat1. EMBO J.
20, 2844–2856
63 Korsisaari, N. et al. (2002) Conditional ablation of the Mat1 subunit of
TFIIH in Schwann cells provides evidence that Mat1 is not required
for general transcription. J. Cell Sci. 115, 4275–4284
64 Li, T. et al. (2004) Failure to proliferate and mitotic arrest of
CDK11p110/p58-null mutant mice at the blastocyst stage of embryonic
cell development. Mol. Cell. Biol. 24, 3188–3197
65 Ortega, S. et al. (2002) Cyclin D-dependent kinases, INK4 inhibitors
and cancer. Biochim. Biophys. Acta 1602, 73–87
Review
TRENDS in Biochemical Sciences
66 Wolfel, T. et al. (1995) A p16Ink4a-insensitive Cdk4 mutant targeted
by cytolytic T lymphocytes in a human melanoma. Science 269,
1281–1284
67 Corcoran, M.M. et al. (1999) Dysregulation of cyclin dependent kinase
6 expression in splenic marginal zone lymphoma through chromosome
7q translocations. Oncogene 18, 6271–6277
68 Hayette, S. et al. (2003) In B-cell chronic lymphocytic leukemias, 7q21
translocations lead to overexpression of the CDK6 gene. Blood 102,
1549–1550
69 Sotillo, R. et al. (2001) Wide spectrum of tumors in knock in mice
carrying a Cdk4 protein insensitive to INK4 inhibitors. EMBO J. 20,
6637–6647
70 Sotillo, R. et al. (2001) Invasive melanoma in Cdk4 targeted mice.
Proc. Natl. Acad. Sci. U. S. A. 98, 13312–13317
71 Knockaert, M. et al. (2002) Pharmacological inhibitors of cyclindependent kinases. Trends Pharmacol. Sci. 23, 417–425
72 Yu, Q. et al. (2001) Specific protection against breast cancers by cyclin
D1 ablation. Nature 411, 1017–1021
73 Zou, X. et al. (2002) Cdk4 disruption renders primary mouse cells
resistant to oncogenic transformation, leading to Arf/p53-independent
senescence. Genes Dev. 16, 2923–2934
74 Miliani de Marval, P.L. et al. (2004) Lack of cyclin-dependent kinase 4
inhibits c-myc tumorigenic activities in epithelial tissues. Mol. Cell.
Biol. 24, 7538–7547
75 Tetsu, O. and McCormick, F. (2003) Proliferation of cancer cells
despite CDK2 inhibition. Cancer Cell 3, 233–245
76 Bai, C. et al. (1994) Human cyclin F. EMBO J. 13, 6087–6098
77 Kong, M. et al. (2000) Cyclin F regulates the nuclear localization of
cyclin B1 through a cyclin–cyclin interaction. EMBO J. 19, 1378–1388
78 Tetzlaff, M.T. et al. (2004) Cyclin F disruption compromises placental
development and affects normal cell cycle execution. Mol. Cell. Biol.
24, 2487–2498
Vol.30 No.11 November 2005
641
79 Chen, X. (2002) Cyclin G: a regulator of the p53–Mdm2 network. Dev.
Cell 2, 518–519
80 Kimura, S.H. et al. (2001) Cyclin G1 is involved in G2/M arrest in
response to DNA damage and in growth control after damage recovery.
Oncogene 20, 3290–3300
81 Zhang, L. et al. (2004) Serine/threonine kinase cyclin G-associated
kinase regulates epidermal growth factor receptor signaling. Proc.
Natl. Acad. Sci. U. S. A. 101, 10296–10301
82 Nakamura, T. et al. (1995) Cyclin I: a new cyclin encoded by a gene
isolated from human brain. Exp. Cell Res. 221, 534–542
83 Kolonin, M.G. and Finley, R.L., Jr. (2000) A role for cyclin J in the
rapid nuclear division cycles of early Drosophila embryogenesis. Dev.
Biol. 227, 661–672
84 Wang, C.Y. et al. (2003) Molecular cloning and characterization of a
novel gene family of four ancient conserved domain proteins (ACDP).
Gene 306, 37–44
85 Krokan, H.E. et al. (2002) Uracil in DNA – occurrence, consequences
and repair. Oncogene 21, 8935–8948
86 Edelheit, S. and Meiri, N. (2004) Cyclin S: a new member of the cyclin
family plays a role in long-term memory. Eur. J. Neurosci. 19, 365–375
87 Malumbres, M. (2005) Revisiting the ‘Cdk-centric’ view of the
mammalian cell cycle. Cell Cycle 4, 206–210
88 Pavletich, N.P. (1999) Mechanisms of cyclin-dependent kinase
regulation: structures of Cdks, their cyclin activators, and Cip and
INK4 inhibitors. J. Mol. Biol. 287, 821–828
89 Sugimoto, M. et al. (2002) Activation of cyclin D1-kinase in murine
fibroblasts lacking both p21Cip1 and p27Kip1. Oncogene 21, 8067–8074
90 Bagui, T.K. et al. (2003) P27Kip1 and p21Cip1 are not required for
the formation of active D cyclin–cdk4 complexes. Mol. Cell. Biol. 23,
7285–7290
91 Thompson, J.D. et al. (1997) The ClustalX windows interface: flexible
strategies for multiple sequence alignment aided by quality analysis
tools. Nucleic Acids Res. 24, 4876–4882
ScienceDirect collection reaches six million full-text articles
Elsevier recently announced that six million articles are now available on its premier electronic platform, ScienceDirect. This
milestone in electronic scientific, technical and medical publishing means that researchers around the globe will be able to access
an unsurpassed volume of information from the convenience of their desktop.
ScienceDirect’s extensive and unique full-text collection covers over 1900 journals, including titles such as The Lancet, Cell,
Tetrahedron and the full suite of Trends and Current Opinion journals. With ScienceDirect, the research process is enhanced with
unsurpassed searching and linking functionality, all on a single, intuitive interface.
The rapid growth of the ScienceDirect collection is due to the integration of several prestigious publications as well as ongoing
addition to the Backfiles – heritage collections in a number of disciplines. The latest step in this ambitious project to digitize all of
Elsevier’s journals back to volume one, issue one, is the addition of the highly cited Cell Press journal collection on ScienceDirect.
Also available online for the first time are six Cell titles’ long-awaited Backfiles, containing more than 12,000 articles highlighting
important historic developments in the field of life sciences.
The six-millionth article loaded onto ScienceDirect entitled "Gene Switching and the Stability of Odorant Receptor Gene Choice"
was authored by Benjamin M. Shykind and colleagues from the Dept. of Biochemistry and Molecular Biophysics and Howard
Hughes Medical Institute, College of Physicians and Surgeons at Columbia University. The article appears in the 11 June issue of
Elsevier’s leading journal Cell, Volume 117, Issue 6, pages 801–815.
www.sciencedirect.com
www.sciencedirect.com