0163-769X/99/$03.00/0
Endocrine Reviews 20(4): 501–534
Copyright © 1999 by The Endocrine Society
Printed in U.S.A.
The Cyclins and Cyclin-Dependent Kinase Inhibitors in
Hormonal Regulation of Proliferation and
Differentiation*
RICHARD G. PESTELL, CHRIS ALBANESE, ANNE T. REUTENS,
JEFFREY E. SEGALL, RICHARD J. LEE, AND ANDREW ARNOLD
The Albert Einstein Cancer Center (R.G.P., C.A., A.T.R., R.J.L.), Department of Developmental and
Molecular Biology and Department of Medicine, Department of Anatomy and Structural Biology
(J.E.S.), Albert Einstein College of Medicine, Bronx, New York 10461; and Center for Molecular
Medicine (A.A.), and Division of Endocrinology and Metabolism, University of Connecticut Health
Center, Farmington, Connecticut 06030
scribed the importance of the coordinated regulatory interactions between components of the cell cycle-regulatory
apparatus and their aberrations in tumorigenesis (1–7). More
recently the mechanisms by which hormone signals affect the
cell cycle-regulatory apparatus have been investigated.
These studies have provided important new insights into the
mechanisms by which hormones regulate diverse processes
including differentiation, proliferation, and apoptosis. This
review describes the role of the cell cycle-regulatory apparatus in hormone-regulated alterations in cellular phenotype.
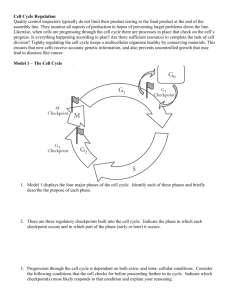
The normal mammalian cell cycle consists of several temporally distinct phases (Fig. 1). One current model of the cell
cycle envisages transitions between different cell cycle states
by passage through checkpoints (1–3, 5, 8) (Fig. 1). Examples
of these states are the initiation and completion of DNA
replication (S) phase and of cell division or mitosis (M).
Between these phases are gaps (G). One important checkpoint in mammalian cells is the restriction point in late G1,
also known as START in yeast. This is the point at which the
cell commits itself irrevocably to another round of DNA
replication. Passage through the restriction point is promoted by a group of G1 cyclins, which include in mid-G1, the
D type cyclins, and in late G1, cyclin E. These cyclins can
heterodimerize with specific catalytic subunits, the cyclindependent kinases (Cdks), to form holoenzymes. Some substrates of these holoenzymes, which are inactivated upon
phosphorylation, are the retinoblastoma tumor suppressor
protein, pRB (retinoblastoma protein) (Fig. 2B) and the related proteins, p130 and p107. It is thought that phosphorylation and inactivation of pRB leads to progression through
the restriction point. The ability of the cyclin/Cdk holoenzymes to phosphorylate pRB is inhibited by a family of small
molecular weight proteins, known as cyclin-dependent kinase inhibitors (CKIs) (Fig. 2A).
Hormonal effects on cell cycle-regulatory proteins can be
considered to occur at several different levels. First, the abundance of both the cyclins and the CKIs can be directly regulated by hormones. Hormones may also directly alter the
subcellular distribution of the cyclins/Cdks or change the
subunit composition of the cyclin/Cdk complexes. Alteration in the composition of the cyclin/Cdk complexes can, in
I. Introduction
II. Regulation of the Cell Cycle G1 Phase
A. The cyclins
B. The Cdk-inhibitory proteins
C. Regulation of transcription by cell cycle-control proteins
III. Endocrine Regulation of Cyclin and Cdk-Regulatory
Proteins
A. Single-transmembrane segment receptors
B. Seven-transmembrane receptors
C. Steroid hormones
D. Intracellular second messengers
IV. Cyclins and CKIs in Endocrine Tumors
A. Parathyroid adenomas
B. Thyroid cancer
C. Pituitary tumors
D. Adrenal tumors
E. Ovarian and testicular cancer
F. Mechanisms of oncogenic transformation by cyclin D1
G. CKIs in treatment of endocrine disease
V. Transcription Factors Regulating the Cell Cycle
VI. Conclusion
I. Introduction
T
HE LAST decade has seen an enormous expansion in our
understanding of the molecular mechanisms governing cell cycle regulation. Several recent reviews have deAddress reprint requests to: Richard G. Pestell, M.D., Ph.D., Departments of Developmental and Molecular Biology and Medicine, Albert
Einstein College of Medicine, Chanin 302A, 1300 Morris Park, Bronx,
New York 10461 USA. E-mail: pestell@aecom.yu.edu
* This work was supported in part by NIH Grants 1R29CA-70897– 01,
R01CA-75503, P50-HL-56399 (to R.G.P.). R.G.P. is a recipient of the Irma
T. Hirschl award and an award from the Susan G. Komen Breast Cancer
Foundation and Mortimer Harrison Foundation. Work at the Albert
Einstein College of Medicine was supported by Cancer Center Core NIH
Grant 5-P30-CA13330 –26 (R.G.P.). A.T.R. was supported by a P. F.
Sabotka Postgraduate Scholarship from the University of Western Australia. J.E.S. is an Established Scientist of the New York City Affiliate
of the American Heart Association and was supported by Grant
USAMRDC-2466. A.A. was supported by NIH Grant RO1 CA-55909.
501
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
502
PESTELL ET AL.
Vol. 20, No. 4
FIG. 1. Schematic representation of the mammalian cell cycle. Competence factors such as PDGF and FGF promote entry into the early
G1 phase. Sequential treatment with progression factors, IGF or EGF,
promote progression through the G1 phase restriction point. Competence of the cyclin D1/Cdk4 complex is induced by mitogens. The
cyclin D1/Cdk4 complex phosphorylates the pRB protein leading to
sequential phosphorylation by cyclin E/Cdk2 and release of free E2F.
The phosphorylation of pRB and relief of transcriptional repression
by pRB induces genes involved in the induction of S-phase entry.
turn, alter their substrate activity. The induction of cyclin/
Cdk abundance can directly affect gene expression through
altering the activity and/or DNA-binding properties of specific transcription factors and the function of the basal transcription apparatus. Thus, complex interactions are induced
as functional transcriptional cascades in part transduced by
the induction of the expression of the cyclin/Cdk proteins.
The coordinated regulation of cell cycle gene expression is
quite specific for a given hormone signal, and specific
changes in abundance of these proteins are required for
hormone signaling to take place.
II. Regulation of the Cell Cycle G1 Phase
A. The cyclins
The cyclin D1 protein is a regulatory subunit of a holoenzyme that phosphorylates and inactivates the tumor suppressor pRB (5, 9, 10). The catalytic subunits, Cdk4 and Cdk6,
are the primary heterodimeric partners of cyclin D1 (1). Overexpression of cyclin D1 acts in G1 to promote progression
through the cell cycle in mammalian cells. Immunoneutralization and antisense experiments have demonstrated that
the abundance of cyclin D1 is rate-limiting in serum-induced
G1 phase progression of Rat1 fibroblasts and MCF7 human
breast cancer cell lines (11–18). Cyclin D1 is not required for
G1-phase progression in cultured mammalian cells that lack
functional pRB (19). In Drosophila melanogaster, cyclin D is
also necessary, but not sufficient, for the initiation of events
leading to S phase entry (20, 21).
The human cyclin D1 gene was cloned as an endocrine
FIG. 2. Cdk Inhibitors. A, Schematic representation of the CKI proteins. The p21 protein contains an amino-terminal Cdk-inhibitory
domain, a nuclear localization signal (NLS) that lies carboxy terminal
to a putative caspase cleavage site (DVHD112L) indicated by an asterisk (474), and a C-terminal domain that binds to PCNA. Although
cyclin A or cyclin E bind only the amino terminus, two distinct cyclinbinding motifs [Cy1 and Cy2 (87)], which bind cyclin E/Cdk2 or cyclin
A/Cdk2 complexes are indicated. Cyclin D1 can bind to the amino
terminus, either alone or in the presence of Cdk4 and binds independently of the K region (87). B, CKI function. The cyclin D1 gene
product binds its catalytic subunit partner (Cdk4) in the presence of
an assembly factor. The cyclin D1/Cdk4 holoenzyme is phosphorylated by a Cdk-activating kinase (475) (CAK), which consists of several subunits. The Ink4 proteins, shown as p16Ink4a, can compete with
D cyclins to form independent binary complexes with Cdk4 (476),
thereby inhibiting the activity of the cyclin D1/Cdk4 holoenzyme. The
role of the p21 CKI family, shown as p27Kip1, in regulating activity of
the cyclin D1/Cdk complex is controversial. In some circumstances
p27Kip1 is thought to inhibit activity of the complex (75). In other
circumstances, p27Kip1 does not inhibit activity of the complex (92)
and acts as an assembly factor (80). These findings suggest that the
stoichiometry or cell type may be important in the action of p27Kip1.
tumor oncogene in human parathyroid adenomas. During
structural analysis of the PTH gene, a pericentromeric inversion was observed in chromosome 11 (22) (Fig. 3A). Cloning and sequencing of the chromosomal breakpoint in these
tumors revealed the presence of an overexpressed gene,
downstream of the PTH gene promoter (22). The PRAD1
cDNA (parathyroid adenoma 1) revealed structural homology to a class of proteins previously identified in yeast
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
FIG. 3. A, Structural organization of the PTH gene in a parathyroid
adenoma demonstrates the pericentric inversion of chromosome 11.
The cyclin D1 gene is positioned 39 to the parathyroid (PTH) generegulatory region. B, The cyclin D1 protein is encoded by five exons.
Conserved motifs include a cyclin box, which makes contact with the
Cdk. This site is conserved between the cyclins. In cyclin A the crystal
structure revealed two sequential 90-amino acid repeats, referred to
as cyclin folds, separated by a short linker peptide. The first fold
corresponds to the cyclin box. An LXCXE motif conserved with several
viral oncogenes is shown, and a site capable of interacting with coactivator proteins (LLESSL) is indicated. The carboxy terminus of
cyclin D1 interacts with HLH proteins involved in myogenesis.
known as cyclins (23). Cyclins were known to regulate the
first Gap phase (G1) in nonmammalian cells. The PRAD1
gene product was thus the first putative mammalian G1
cyclin cloned. Furthermore, the PRAD1 cDNA was capable
of rescuing a yeast mutant in its G1 CLN cyclins, which
suggested the PRAD1 gene may encode a functional cyclin
now called a D-type cyclin (24). Independently, the murine
homolog of PRAD1 was cloned as a colony-stimulating factor-1 (CSF-1)-responsive gene product (25).
PRAD1 or cyclin D1 is expressed relatively ubiquitously
with the exception of normal lymphoid and myeloid cells
(26). The two major transcripts of 4.2- 4.8 kb and 1.3–1.7 kb
detected by Northern blot analysis differ in the length of their
39-untranslated region. Cyclin D1 mRNA levels are induced
during G1 phase progression in most cell types in culture.
The induction of cyclin D1 mRNA by serum or growth factors is rapid, and cyclin D1 is actively degraded upon the
withdrawal of growth factors. The degradation of cyclin D1
protein early in G1 abolishes cell cycle progression, but destruction late in G1 does not.
The 35-kDa cyclin D1 protein is encoded by 5 exons in the
structurally similar human and mouse genes (26). The cyclin
D1 protein shares structural homology to the other cyclins.
The amino terminus of cyclin D1 contains a motif Leu-X-
503
Cys-X-Glu (where X represents any amino acid). This motif,
which is shared by the viral oncoproteins E1A, simian virus
(SV) 40 large T antigen, and papillomavirus E7, is involved
in binding the pRB pocket domain (Fig. 3B). A region located
carboxy terminal to the pRB-binding domain is known as the
“cyclin box” because it is conserved between the known
cyclins. The acidic rich carboxy terminus of cyclin D1 inhibits
myogenic helix loop helix (HLH) protein function (27). An
alternate splice form of cyclin D1 encodes a protein with an
altered carboxy-terminal domain (CTD) (28). The cyclin D1
protein is quite unstable, with a half-life of less than 20 min,
with degradation occurring through ubiquitin proteosomemediated degradation (29). In quiescent cells, cyclin D1 protein levels are low. However, nuclear abundance increases as
cells progress through G1 phase (11). As cells pass into S
phase, cyclin D1 moves from the nucleus to the cytoplasm.
Exclusion of cyclin D1 from the nucleus is required for progression into S phase in human fibroblasts (11). Proliferating
cell nuclear antigen (PCNA), which is required for DNA
polymerase d activity, binds cyclin D1 in the nucleus. As cells
enter S phase, cyclin D1 protein no longer binds PCNA and
is extruded from the nucleus (30).
Two other structurally related cyclins, D2 and D3, are also
capable of heterodimerizing with Cdk4/6 and phosphorylating pRB in vitro. The amino acid identity between the
human D-type cyclins is 53– 63%. Cyclin D2 was mapped to
chromosome 12 at 12p13 and cyclin D3 was mapped to 6p21.
It is currently thought that each of the D-type cyclins subserve multiple functions, some of which are shared, such as
phosphorylation of the pRB protein, and some of which are
distinct. The mRNA distribution and expression profiles differ between the D-type cyclins. Steady state cyclin D2 mRNA
levels, for example, peaked in late G1 (31) and for cyclin D3
peaked in S phase rather than early in G1 as seen for cyclin
D1. Unlike the relatively ubiquitous distribution of cyclin D1
expression, the expression of cyclin D2 is somewhat more
restricted with mRNA expressed abundantly in T lymphocytes and gonadal cells, although several different transformed cell lines also express cyclin D2 (25, 31–33).
Cyclin E was first identified by screening human cDNA
libraries for genes that would complement G1 cyclin mutations in Saccharomyces cerevisiae and has subsequently been
found to have specific biochemical and physiological properties that are consistent with a G1 function in mammalian
cells. mRNA levels for cyclin E peak later in G1 phase than
cyclin D1. Mammalian cells express several isoforms of cyclin E protein. These proteins are encoded by alternatively
spliced mRNAs and are localized to the nucleus during late
G1 and early S phase (34). The cyclin E-Cdk2 complex is
maximally active at G1/S, and overexpression of cyclin E
decreases the time it takes the cell to complete G1 and enter
S phase. The destruction of cyclin E is linked to ubiquitinmediated degradation. Cyclin E phosphorylation is coupled
to cyclin E turnover via site-specific phosphorylation, which
acts as a signal for ubiquitination and proteasome processing
(35, 36).
Cyclin D1 and cyclin E appear to subserve at least partially
overlapping functions. pRB is phosphorylated at an overlapping subset of sites by these two distinct kinases (37).
Several different scenarios may explain why two different
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
504
PESTELL ET AL.
kinases subserve this function. Phosphorylation of pRB at
distinct sites may alter the ability of pRB to interact with a
distinct subset of transcription factors or substrates (38). Alternatively, phosphorylation at distinct sites by both kinases
may alter the epitopes of pRB required for full phosphorylation. It is clear, however, that the effect of cyclin D1 to
promote cell cycle progression in cultured cells requires pRB,
while the effect of cyclin E occurs independently of pRB (19,
39, 40). Thus, fibroblasts engineered to constitutively overexpress cyclin E showed elevated cyclin E-dependent kinase
activity and a shortened G1 phase of the cell cycle (40, 41).
Under certain circumstances, in cultured Rat-1 cells, cyclin
D1, but not cyclin E, induced pRB phosphorylation, suggesting cyclin D1 and cyclin E promote G1 phase progression
through different mechanisms (42). However, recent studies
in NIH3T3 cells demonstrated that cyclin E can phosphorylate pRB and located a pRB-binding motif VxCxE (43). Mutation of the pRB-binding motif abolished the ability of cyclin
E to promote S phase entry in NIH3T3 cells (43). It is clear
that, in addition to pRB, alternate substrates for cyclin
D/Cdk exist (below). Histone H1 is also phosphorylated well
by cyclin E/Cdk2. Because cyclin E enhancement of G1 phase
progression occurs independently of pRB (14, 40), a search
has begun for alternate cyclin E/Cdk substrates. Recent studies have identified one new substrate for cyclin E/Cdk2,
known as the NPAT protein (nuclear protein mapped to the
ATM locus) (44). It is thought that phosphorylation of NPAT
by cyclin E/Cdk2 may promote S phase entry.
Cyclin A plays an essential role in the progression through
the cell cycle as a regulatory component of the Cdk2 and cdc2
kinases (1, 45). The mRNA and protein of cyclin A accumulate at the end of the G1 phase. Microinjection of immunoneutralizing antibodies to cyclin A or antisense expression
vectors indicate a requirement for cyclin A in DNA replication (46). Complexed to Cdk2 in cooperation with cyclin E,
cyclin A has been shown to trigger S phase entry of G1 nuclei
from HeLa cells (47). Cyclin A has been shown to promote
S phase entry by associating with transcription factors and by
regulating target genes involved in cell growth regulation.
In normal human fibroblasts, cyclin A/Cdk2 forms a quaternary complex with p21Cip1 and PCNA and cyclin A forms
associations with p107 and E2F transcription factors. Cyclin
A/Cdk2 binds to the amino terminus of the E2F-1–3 transcription factors. The E2F multigene family contains six
members, and preferential binding of cyclin A/Cdk2 to E2F1–3 likely alters the repertoire of E2F transcriptional target
genes. The E2F proteins bind to DNA with an heterodimeric
binding partner from the DP protein family and activate gene
transcription through the E2F enhancer sequence. This DNAbinding site was initially identified as an adenovirus AdE2a
enhancer sequence and has since been identified in the promoter of a number of genes induced during G1/S phase
transition. E2F-1 is phosphorylated by cyclin A/Cdk2,
down-regulating its ability to bind DNA and activate transcription (48 –50). The region of E2F-1 phosphorylated by
cyclin A/Cdk2 is conserved among E2F proteins (E2F-1,
E2F-2, -E2F-3), implying each of the protein’s binding activity
may be regulated in this manner. E2F-1 mutants defective in
cyclin A binding cause apoptosis and deregulated cell cycle
Vol. 20, No. 4
progression, indicating the importance of E2F-1 phosphorylation-dependent inactivation (49, 50).
A number of other cyclins and Cdks have been identified
and are touched on only briefly herein. The abundance of
cyclin F fluctuates during cell cycle progression like cyclin A,
peaking in G2 (51). Overexpression of cyclin F increases the
proportion of cells in G2 phase (51). The expression of cyclin
G (52) is induced upon nerve injury in the motor neurons
during the early phase of the nerve regeneration process (53).
A neuron-specific cyclin p35 has been described that is expressed in postmitotic neurons of the central nervous system
(CNS) (54, 55). The heterodimeric partner for p35 is Cdk5 (55,
56). Mice lacking p35 develop abnormal lamination likely
due to abnormal neuronal migration (57). The related cyclin
p39 is also required for neurite outgrowth in a cultured
hippocampal cell line model (58). Cyclin H and Cdk7 form
a Cdk-activating kinase (CAK), which regulates activities of
the cyclin/Cdk complexes and activity of the RNA Pol II
CTD. Unlike many of the other cyclins the abundance of CAK
does not change during cell cycle progression, although its
activity and substrates may (discussed below). Mitotic entry
is signaled by the accumulation of cyclin B-cdc2 (59). Although accumulating in S and G2 phases, the activity of cyclin
B/cdc2 is inhibited by phosphorylation on Tyr-15 and Thr-14
by Wee1/Mik1/Myt1-related protein kinases. The Cdc25c
phosphatase is stimulated to dephosphorylate T14/Y15 and
activate Cdc2. Destruction of the B-type cyclins through a
ubiquitin-dependent proteolysis is required for progression
past anaphase (60, 61).
Thus, specific cyclins convey cell-type dependent developmental functions and regulate specific steps in progression
through the cell cycle of dividing cells. Recent attention has
been drawn to aberrations in function of these proteins in
tumorigenesis. The normal function of these cyclins in the
mature animal in nondividing cells, however, remains to be
fully understood.
B. The Cdk-inhibitory proteins
Activity of the holoenzyme containing cyclin D1 (cyclin
D1/Cdk4 and cyclin D1/Cdk6) referred to here as cyclin D1
kinase (CD1K), is modulated in vitro by the Cdk-regulatory
proteins (62), which are divided into two broad categories
(Fig. 2). The first family, the Ink4s (p16Ink4a, p15Ink4b, p18Ink4c,
and p19Ink4d), inhibit specifically Cdk4 or Cdk6 and contain
a set of highly conserved ankyrin ring motifs (Fig. 2A). The
second group include the Cip/KIP family (p21Cip1, p27Kip1,
and p57Kip2), which share partial structural homology and
possess the ability to inhibit cyclin/Cdk complexes. All CKIs
can cause G1 arrest when overexpressed in transfected cells
(reviewed in Ref. 2). The expression and subcellular distribution of the CKIs is regulated by hormones in a complex and
cell-type specific manner (Fig. 2).
Overexpression of Ink4 proteins in vitro dissociates the
CD1K complexes and overexpression of p16Ink4a is associated
with reduced levels of cyclin D1-holoenzyme complexes
(Fig. 2B). The p16 (INK4a/ARF) locus encodes two alternate
transcripts, the p16Ink4a and the p19ARF (alternate reading
frame). Overexpression of either of these transcripts can induce cell cycle arrest and block transformation (63– 65).
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
Transgenic mice homozygously deleted of the p16 locus
spontaneously developed a variety of malignancies including lymphomas and fibrosarcomas (66), and deletion of the
p19ARF gene resulted in animals with a similar phenotype
(67). Unlike p16Ink4a, however, the cell cycle arrest induced
by p19ARF is p53 dependent. p19ARF directly binds MDM2
and stabilizes p53 (65, 68). The human equivalent to p19ARF,
p14ARF, also binds MDM2, resulting in stabilization of both
p53 and MDM2 (69) (Fig. 2B). Consistent with a negative
feedback loop, p53 in turn inhibits p14ARF (69) and p19ARF
expression (70). Unlike p53, however, p19ARF is not involved
in the DNA damage response (69). Together these studies
suggest that the alternate reading frames of the CdkN2A locus
function to inhibit cell cycle progression through two different mechanisms (71). The evolutionary pressure to sustain
the presence of these two tumor suppressor genes, possibly
as the result of gene duplication, in such close proximity and
given their vulnerability to co-deletion, remains an area of
considerable speculation. The possibility that each transcript
plays a role in hormonal signal transduction specificity has
yet to be explored.
The second class of inhibitors, the p21 family, includes
p21Cip1 (72, 73), p27Kip1 (74, 75), and p57Kip2 (76 –78). Although intially referred to as CKIs, with biochemical properties supporting this notion (79), strong evidence is accumulating that p21Cip1 and p27Kip1 function as assembly
factors in vitro (80) and in vivo (81) to enhance cyclin Ddependent activity. The p21 family members have a conserved region near the amino terminus that is necessary and
sufficient for binding to and inhibiting Cdk2 (82– 84). At
certain concentrations, however, p21Cip1 does not inhibit
cdc2 or Cdk2 kinase activity (85). Thus although p21Cip1
inhibits Cdk4 and Cdk6 kinase activity, with an inhibition
constant (Ki) of 0.5–15 nm, p21Cip1 is a poor inhibitor of
cdc2/cyclin B in vitro with a Ki of 400 nm (86). In subsequent
studies, functional subdomains of p21Cip1 were defined with
fine deletional analysis. For example, the binding of p21Cip1
to cyclin E/Cdk2 and cyclin A/Cdk2 was shown to involve
both a Cdk2-binding domain and either an amino-terminal
or carboxy-terminal cyclin-binding domain whereas binding
by cyclin D1 involved only the amino-terminal cyclin-binding domain (87) (Fig. 2A). The carboxy-terminal region of
p21Cip1 allows it to associate with PCNA, a processivity subunit of the DNA polymerase d holoenzyme (82– 84, 88, 89).
Because the binding of p21Cip1 to PCNA inhibits the processivity of polymerization, but does not affect excision repair,
it was suggested that p21Cip1 may serve to coordinate DNA
replication with cell cycle progression (90).
The p212/2 embryonic fibroblasts derived from mice homozygously deleted of the p21Cip1 gene (91) exhibit a significant growth alteration in vitro, achieving a saturation
density as high as that observed in p532/2 cells. In addition,
p212/2 cells are significantly deficient in their ability to arrest
in G1 in response to DNA damage and nucleotide pool perturbation. Although the p21 family of proteins inhibit the
activity of Cdk2-containing complexes, recent studies demonstrated that the p21 family may also function under some
circumstances as assembly factors to promote the association
of Cdk4 with D-type cyclins (80, 92). The p21 family proteins
enhance activity of the cyclin D1-kinase (CD1K) complex at
505
low concentrations, while inhibiting CD1K complex at high
concentrations.
The p27Kip1 protein was initially characterized as a protein
homologous to the tumor suppressor p21Cip1. When overexpressed in fibroblasts, cell cycle progression was delayed
and antisense p27Kip1 experiments resulted in mitogenindependent G1 phase progression, indicating a critical role
for p27Kip1 in the establishment or maintenance of cellular
quiescence (93, 94). The abundance of p27Kip1 is regulated
primarily at a posttranslational level, although translational
control also contributes. Thus, p27Kip1 mRNA levels remain
relatively unchanged during the cell cycle transition; however, the addition of mitogens reduces p27Kip1 protein levels.
For example, in quiescent 3T3 cells p27Kip1 protein levels
decrease after mitogenic stimulation (95–97). The growth
factor-mediated reduction in p27Kip1 protein levels is mediated primarily through enhanced ubiquitin-mediated degradation (98). It will be of interest to determine whether
progression through the G1/S phase of the cell cycle is associated with induced expression of proteolytic enzymes,
such as members of the caspase family. p27Kip1 abundance
was also implicated as an important mediator of the cytostatic effects of rapamycin and cAMP (75, 99, 100), although
these implications were not borne out in all studies using
transgenic mice models (below).
The cyclin/Cdk complex to which p27Kip1 is bound determines its functional activity. p27Kip1 is found associated
with cyclin E in a variety of cell types during quiescence (95,
99). When bound to cyclin D1/Cdk4, p27Kip1 may not be
inhibitory (74, 99, 101, 102), whereas cyclin E/Cdk2 activity
is inhibited by p27Kip1. It is thought that the removal of
p27Kip1 from the cyclin E/Cdk complex is an essential step
for S-phase entry. Through binding cyclin D1/Cdk4, p27Kip1
is sequestered from cyclin E/Cdk2, reducing its inhibition by
p27Kip1 (74, 99, 101, 102).
The degradation of p27Kip1 upon mitogen stimulation is
dependent upon prior phosphorylation. Expression of cyclin
E/Cdk2 in murine fibroblasts was found to induce phosphorylation of p27Kip1 on T187 (103). A mutant of p27Kip1 at
amino acid T187, to alanine, created a p27Kip1 protein that
caused a G1 block resistant to cyclin E overexpression and
whose level of expression was not modulated by cyclin E.
Phosphorylation of p27Kip1 by cyclin E/Cdk2, enhanced degradation of p27Kip1, thereby promoting G1-S phase transition.
Thus, the cyclin/Cdk complexes promote cell cycle progression in mammalian cells by also enhancing degradation of
the CKI.
Together these studies suggest that p27Kip1 interacts with
cyclin E/Cdk2 in two distinct ways. First, through tightly
binding to cyclin E/Cdk2, p27Kip1 inhibits the ability of the
holoenzyme to phosphorylate target substrates, such as pRB,
or the NPAT protein (44). Second, p27Kip1 is phosphorylated
by cyclin E/Cdk2, leading to the release and subsequent
degradation of p27Kip1. The ability of p27Kip1 to either block
Cdk activity or serve as a substrate for the Cdk appears to be
determined by the ambient concentration of ATP within the
reaction. At low ATP concentrations (,50 mm) p27Kip1 is
primarily a CKI, but at ATP concentrations approaching
physiological levels (.1 mm), p27Kip1 is more likely to be a
substrate.
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
506
PESTELL ET AL.
The homozygous deletion of the p27Kip1 gene resulted in
animals with organomegaly, intermediate lobe pituitary tumors, and testicular and ovarian cell hyperplasia (104, 105).
Because p27Kip1 expression was induced upon oligodendrocyte differentiation, the role of p27Kip1 in this process was
closely examined in mice homozygously deleted of the
p27Kip1 gene [p27 knockout (KO)]. The oligodendrocytes derived from p27 KO mice underwent prolonged proliferation
and delayed differentiation, suggesting a role for p27Kip1 in
promoting oligodendrocyte proliferation (106). In view of
previous studies demonstrating the induction of p27Kip1 by
transforming growth factor-b (TGFb) and rapamycin, signaling by these agents was assessed in the p27 KO mice.
Surprisingly, initial analysis of p27Kip1 KO T cells suggested
their proliferation rates in response to TGFb and rapamycin
were similar to those of wild type, suggesting that p27Kip1
was not required for the cytostatic effect of these two agents
(104, 105). In subsequent experiments, however, exponentially growing p272/2 fibroblasts were found to have an
impaired antiproliferative response to rapamycin. The inhibition of DNA synthesis by rapamycin was approximately
half of control p271/1 fibroblasts (107). In addition, the
p272/2 T lymphocytes were 15- to 30-fold more sensitive to
the growth-inhibitory effect of rapamycin (107). The different
results obtained by these investigators may be due to methodological differences, but together provide support for the
role of p27 in rapamycin-dependent inhibition of cellular
proliferation. The p27Kip1 gene product is not a significant
source of specific selected inactivating mutations, however,
raising suspicions that this locus may not encode a human
tumor suppressor gene. The results of studies exposing
p27Kip1 KO mice to carcinogenic agents or matings to transgenic tumor-prone animals may provide important insights
into the potential tumor suppressor function of the p27Kip1
gene product.
p57Kip2 was also cloned as a protein related to p21Cip1 and
p27Kip1. Overexpression of p57Kip2 was found to arrest cells
in G1 (77). Both the amino-terminal cyclin/Cdk binding domain and the carboxy-terminal PCNA binding domains of
p57Kip2 were required for full antimitogenic activity and
inhibition of cellular transformation much like p21Cip1 (78).
Unlike p21Cip1, however, p57Kip2 was not regulated by p53.
Homozygous deletion of the murine p57Kip2 allele by one
group of investigators resulted in animals with abnormal
endochondral ossification (108), while other investigators
observed increased prenatal growth, adrenal cortical hyperplasia, and cytomegaly (109), features found in patients with
Beckwith-Wiedemann syndrome. The pituitary tumors, together with the testicular and ovarian cell hyperplasia in the
p27Kip1 KO mice and the adrenal cortical hyperplasia and
cytomegaly in the p57Kip2 KO mice, launched an excited
search for a role of these proteins in the regulation of normal
function in these endocrine tissues (below).
Since the original cloning of the CKI proteins, it has become clear that the abundance and activity of these proteins
are regulated in a complex manner by hormonal stimuli.
Frequently the overexpression of the protein may be sufficient to induce cell cycle arrest, but equally frequently the
overexpression of the protein, as observed during differentiation, has been shown to be insufficient to recapitulate the
Vol. 20, No. 4
differentiated phenotype induced by a particular hormone.
Furthermore, in most circumstances, homozygous deletion
of the CKIs has not affected the signaling pathway that induced the abundance of the CKI. Together, these types of
findings have led to an understanding that the precise timing
and orchestration of the alteration in expression of the CKI,
together with changes in their subcellular distribution and
the formation of multimeric complexes in the cell, may be
critical for the induction of the differentiated phenotype.
Thus, although the instruments have been identified, the
mechanisms of orchestration are poorly understood and are
critical for the hormonal induction of the cellular phenotype.
C. Regulation of transcription by cell cycle-control proteins
As noted above, hormonal signals regulate the abundance
of cell cycle-control proteins. The altered levels of the cyclins/Cdks in turn regulate downstream target genes. These
genetic effects are conveyed by altering the activity of transcription factors and/or by regulating the basal transcription
apparatus. Transcription factors that coordinate hormonemediated signal transduction can be regulated by the cyclin/
Cdk complexes through several different mechanisms (Fig.
4). These effects can be thought of as either regulating transactivation function (Fig. 4, A and B), by altering DNA binding
(Fig. 4, C and D), or by altering protein/protein interaction
(Fig. 4, E and F).
Transcription factor binding to DNA can be affected by cell
cycle-control proteins. p53, whose activity has been linked to
transcriptional activity of a number of hormone-regulated
genes, is phosphorylated specifically by cyclin A/Cdk2 and
cyclin B/cdc2, consistent with the timing of p53 phosphorylation in vivo (110). Phosphorylation of p53 on serine 315 by
these cyclin holoenzymes in vitro enhances selectivity of p53
DNA binding activity, in particular to DNA sequences in the
p21Cip1 promoter (Fig. 4A). Another example of this mechanism is the inhibition of the DNA binding affinity of the
E2F/DP transcription factors by cyclin A/Cdk2 (Fig. 4B).
This phosphorylation-mediated inhibition of E2F/DP protein binding may be important in attenuating the transcriptional enhancer activity of these complexes after S phase
entry. Because E2F-1 overexpression is capable of inducing
apoptosis in many cell types, the inhibition of E2F/DP activity is likely important in cell survival.
Cell cycle-regulatory proteins can also affect the transactivation function of specific transcription factors. The transcription factor B-Myb, for example, which regulates activity
of gene expression through the Myb-binding site (PyAACG/
TG), is phosphorylated and activated by cyclin A/Cdk2 (111)
(Fig. 4C). Several nuclear receptors are also phosphorylated
and activated by the cyclin/Cdks, including the glucocorticoid and estrogen receptors (Section III.C below). The glucocorticoid receptor, for example, is phosphorylated by cyclin E/Cdk2 (112). Cell cycle-control proteins can also
regulate gene expression by promoting transcription factor
binding through inhibiting repressors. The HLH protein Id2
resembles the basic HLH proteins that govern gene expression through E box sequences (Fig. 4D). The Id family of
proteins lack basic DNA-binding domains and therefore
function as transdominant negative regulators through E box
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
FIG. 4. Regulation of transcription factors by the Cdks. A, Enhanced
transcription factor binding activity. The phosphorylation of p53 by
cyclin A/Cdk2 (110) enhances the specificity of its binding to target
DNA sequences in the p21Cip1 promoter. B, Reduced transcription
factor binding activity. Phosphorylation of E2F-1 at the amino terminus by cyclin A/Cdk2 inhibits E2F/DP-1 binding activity (477). The
phosphatase PP2A is also capable of enhancing phosphorylation and
thereby reducing DP-1 binding activity (367). The loss of E2F/DP
binding reduces the transcriptional enhancement of target genes containing E2F-binding sites. C, Induction of transcription factor activity. Cyclin A/Cdk2 holoenzymes phosphorylate B-myb to enhance its
transcriptional activity through the myb-binding site (MBS). (D).
Inhibition of transcription repression. The transcriptional repressor
Id2 heterodimerizes with basic HLH proteins that normally activate
gene transcription through E box sequences. As Id2 is incapable of
binding DNA, it functions as trans-dominant negative regulator of E
box-mediated transcription. Cyclin E/Cdk2 or cyclin A/Cdk2 phosphorylate and inhibit Id2 function, promoting E box-dependent transcription. E, Protein-protein interactions. Cell cycle-regulatory proteins form protein-protein interactions with known transcription
factors and alter their activity independently of the kinase activity.
For example, cyclin D1 binds to the ER and enhances ERE enhancer
activity (114, 115).
sequences. Phosphorylation of Id2 by cyclin E/Cdk2 or cyclin A/Cdk2 leads to a reduction in the transcriptional repressor function of Id2 and enhances activity through the E
box sequence (113). As noted above, the transcriptional activity of the E2F-1 factor is inhibited by cyclin A/Cdk2 binding and phosphorylation (48 –50).
Cell cycle-regulatory proteins can also affect signaling by
forming direct protein-protein interactions with known tran-
507
scription factors. For example, cyclin D1 can bind the estrogen receptor (ER), enhancing the activity of the ER through
a synthetic estrogen response element (114, 115). Cyclin D1
also binds a Myb-related protein, DMP1, and modulates its
activity (116). Although the nature of the association remains
to be fully defined, cyclin D1 also inhibits v-Myb transcriptional activity independently of Cdk activity (117) and cyclin
D1 interacts with HLH proteins to inhibit their activity, at
least in part, independently of the Cdk domain of cyclin D1
(27). Finally, a growing list of transcription factors are regulated in their activity by either the pRB or pRB-related
proteins. Activity of the Sp1 transcription factor is induced
by the pRB protein (118). Although the mechanisms by which
pRB regulates the activity of these other transcription factors
remain to be fully determined, other transcription factors
capable of binding to and/or regulated by pRB include Elf-1,
E2Fs, PU.1, c-Myc, Sp-1, MyoD, ATF-2, NF-IL6, and UBF
(reviewed in Refs. 119 and 120). For the time being, these
interactions are loosely classified as pRB dependent (Fig. 4E).
Components of the basal transcription apparatus are also
regulated by the cyclin/Cdks (121). The RNA polymerase II
large subunit contains an essential CTD. The CTD is phosphorylated on a fraction of the RNA polymerase II molecules
in vivo and is phosphorylated by the general transcription
factor TFIIH in vitro. TFIIH is composed of at least nine
subunits, which include Cdk7, cyclin H, and MAT1 (p36).
These three subunits (Cdk7, cyclin H, MAT1) form a ternary
complex, Cdk-activating kinase (CAK). The kinase activity of
the TFIIH complex is directed toward the RNA Pol II CTD
(122, 123). CAK preferentially phosphorylates Cdk2,
whereas TFIIH preferentially phosphorylates the CTD (124).
CAK also phosphorylates the threonine residue of the Cdk
when cyclin D1-Cdk has formed a dimeric complex, and
CAK is required for full cyclin D1/Cdk4 activity (1, 125).
Recent studies have demonstrated that the cyclin H/Cdk7
complex preferentially phosphorylated Cdk2, whereas the
multimeric complex that included cyclin H/Cdk7 and
MAT1(p36) preferentially phosphorylated the RNA Pol II
CTD (126). Thus, the substrate specificity of cyclin H/Cdk7,
which is abundant within the cell, is dictated by the presence
of MAT1 (124, 126). The CTD is also phosphorylated by a
complex known as P-TEFb which includes Cdk9 and cyclin
T (which includes T1, T2a, and T2b) (127).
RNA Pol II and Pol III-dependent transcription is reduced
during mitosis in part through cell cycle-regulatory proteins
(128, 129). RNA Pol III activity is increased in pRB-deficient
mice and pRB targets TFIIB. As pRB contains regions of
homology to TATA-binding protein and BRF, components of
TFIIB, it is thought that pRB disrupts TFIIB by mimicking
these two components (130). Mitosis-specific exclusion of
transcription factors from chromatin may also be important
in cell cycle-specific gene regulation. Phosphorylation and
inhibition of poly (A) polymerase by cyclin B/Cdc2 may
contribute to the overall decrease in RNA and protein synthesis that occurs during mitosis (129, 131, 132).
Cell cycle-regulatory proteins also affect the high molecular weight coactivator proteins of the p300/CBP family. The
p300 protein was noted to undergo phosphorylation during
cell cycle transition and during cell cycle exit in myocyte
differentiation (133). These high molecular weight coactiva-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
508
PESTELL ET AL.
tors, (p300/CBP), interact with a broad array of nuclear receptors and general transcription factors, including members
of the AP-1 family, which are involved in transducing hormone signals (134, 135). The general effects of coactivators on
gene transcription are mediated in part through modulating
histone structure either through direct histone acetylation
activity of the transcription coregulator (136) or through the
ability of the coregulator to recruit other proteins with histone-regulatory function (137, 138). The p300/CBP family
also regulate cell cycle-regulatory transcriptional targets including p21Cip1 and cyclin D1 (139 –141). To date, relatively
little is known about the hormonal regulation of these associations, although these interactions provide important evidence for cross-talk between the cyclins and the basal transcription apparatus.
III. Endocrine Regulation of Cyclin and
Cdk-Regulatory Proteins
A. Single-transmembrane segment receptors
1. Protein tyrosine kinase receptors.
a. CSF-1. CSF-1 was the first of a group of factors purified
(142) that stimulate the formation of colonies of mature myeloid cells from immature hemopoietic precursors (143).
Binding of CSF-1, a homodimeric glycoprotein, to its highaffinity receptor, encoded by the c-fms protooncogene, initiates dimerization, autophosphorylation, and signaling
(143, 144). SH2 domain-containing substrates binding to autophosphorylated c-Fms have been implicated in signaling
by CSF-1 and include STAT (signal transducer and activator
of transcription)-1, Grb2, Src, and phosphatidylinositol 3-kinase (PI3-kinase).
As noted above, the murine homolog of PRAD1 was initially cloned as a CSF-1-responsive gene product (25). Many
studies of CSF-1 signaling were performed in the murine
macrophage cell line, BAC1.2F5. In these cells CSF-1 induces
dimerization and autophosphorylation of the CSF-1 receptor,
as well as tyrosine phosphorylation of the p85 subunit of
PI3-kinase, c-Cbl, SHP-1, and Tyk2. CSF-1 induces the activity of STAT proteins, Raf-1, MEK, ERKs (extracellular
signal-regulated kinases), and S6 kinase. Although it is clear
that the expression of c-fos, c-myc, and cyclin D1 are enhanced
by CSF-1, there is controversy as to whether the induction of
c-fos and c-myc both lie downstream of a Ras-signaling pathway (145). Overexpression of oncogenically activated Raf-1
protein in BAC1.2F5 macrophages induced cellular proliferation and immediate early gene expression without induction of ERK activity, demonstrating that ERK signaling was
not required for cellular proliferation (146).
The introduction of ligand-activated human c-Fms receptor can also induce proliferation when ectopically expressed
in other cell types, including NIH-3T3 cells. The use of dominant negative expression vectors in NIH3T3 cells helped
map an important role for a proliferative pathway involving
Ras, Ets proteins, and c-Myc (147). This pathway, although
necessary, was not sufficient for CSF-1 signaling to promote
proliferation (147). In NIH3T3 cells overexpressing a mutant
c-Fms receptor, Y809F, the defective mitogenic signaling and
c-myc expression could be rescued by cyclin D1 overexpres-
Vol. 20, No. 4
sion (148). Studies by Barone and Courtneidge (149) suggested that the induction of DNA synthesis by CSF-1 in
NIH3T3 cells required both a Ras-dependent pathway that
induced Fos and a Ras-independent pathway that induced
c-myc transcription, suggesting that Fos and c-myc may function in parallel pathways.
b. Platelet-derived growth factor (PDGF). The presence of
mitogens in platelets was first suggested by Balk (150) who
observed that serum-supplemented culture medium supported the proliferation of chicken fibroblasts better than
plasma-supplemented medium. Each of the three possible
isoforms of this dimeric peptide, PDGF, made of A and B
chains, have been isolated from natural sources including
human platelets. The most prominent activity of PDGF is to
stimulate proliferation. Growth factors have been categorized as either competence- or progression-inducing agents
(151). Transient exposure of BALB/c3T3 cells to PDGF or
fibroblast growth factor (FGF) induces a state of competence
to undergo DNA synthesis, while insulin-like growth factors
(IGFs), epidermal growth factor (EGF), and other substances
that are present in plasma permit progression through the G1
phase of the cell cycle (152). In BALB/c-3T3 cells, two arbitrary control points were described in the G1 phase: the
V-point, typically occurring 6 h into G1 phase — a point at
which the cell fails to progress further without the addition
of essential nutrients including amino acids, and the W point,
which occurs later at the G1/S border (153).
In keeping with the known effect of PDGF early in G1
phase, PDGF treatment of a variety of cell types was associated with an induction of cyclin D1 abundance (17, 154, 155)
and CD1K activity (17, 155). In primary tracheal myocytes,
PDGF induced cyclin D1 expression with a sequential induction of pRB phosphorylation (17). The induction of S
phase traversal was also blocked by immunoneutralizing
antibodies to cyclin D1 in these cells (17). The induction of
cyclin D1 by PDGF in CHO cells was dependent upon Ras
(155).
PDGF directly affects the abundance and subcellular localization of the CKI, p27Kip1. PDGF has been shown to
reduce p27Kip1 levels in fibroblasts (96); however, the relative
abundance of p27Kip1 in a p27Kip1/cyclin D1/Cdk4 multiprotein complex increased with PDGF treatment of fibroblasts and was associated with increased pRB kinase activity,
consistent with a model in which p27Kip1 binding to specific
partners is important in determining pRB kinase activity
(156). PDGF treatment of BALB/c-3T3 cells resulted in both
a reduction in p27Kip1 protein synthesis (156) and an increase
in p27Kip1 protein degradation. In PDGF-treated BALB/c3T3 cells, p27Kip1 protein associated with cyclin D1 increased
as the abundance of p27Kip1 bound to cyclin E complexes
decreased. The induction of p27Kip1 degradation by PDGF in
Chinese hamster embryo fibroblasts was blocked by dominant negative mutants of Ras and RhoA (155). Because the
induction of cyclin D1 by PDGF was dependent only upon
Ras, these findings suggest distinct Ras family members regulate the effect of PDGF on distinct components of the cell
cycle-regulatory apparatus.
c. Basic FGF (bFGF). bFGF was the first member of a family
of structurally related proteins, isolated as a biochemically
distinct mitogen for 3T3 fibroblasts from pituitary gland
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
crude extracts (157). Together with the 11 other members of
the FGF family (aFGF, int-2, hst, FGF-5, FGF-6, KGF, AIGF,
GAF, FGF-10, FGF-11, and FGF-12), bFGF has been shown to
exert mitogenic and differentiation inducing potential in a
wide variety of cell types. The high-affinity FGF receptors are
single-chain transmembrane proteins of the Ig-like receptor
superfamily. The binding of ligand induces dimerization,
tyrosine transphosphorylation, and phosphorylation and activation of a broad array of substrates including c-Src and the
F-actin-binding protein cortactin, which correlates with the
induction of DNA synthesis (158). The induction of SHC/
Grb2/SOS complex formation with Ras and subsequently
ERK activation has been linked to the induction of DNA
synthesis primarily in cell types that are involved in tissue
repair and revascularization such as fibroblasts, chondrocytes, myoblasts, smooth muscle, and endothelial cells (159).
FGF can induce either proliferation or differentiation depending upon the cell type. It was anticipated, therefore, that
distinct changes in the abundance or composition of cell
cycle-control proteins may mediate these distinct responses.
To examine this possibility, a number of different groups
analyzed cell cycle-control protein abundance in FGF-treated
cells. In fibroblasts, FGF treatment is associated with the
induction of DNA synthesis and the induction of cyclin D1
levels. In MCF7 cells, FGF induces ERK activity with an
initial mitogenic and subsequently a cytostatic signal. Cyclin
D1 protein levels are initially induced in bFGF-treated MCF7
cells, and then increasing p21Cip1 levels contribute to reduced
pRB phosphorylation and the inhibition of cellular proliferation (160). Differentiation of skeletal myoblasts in culture is
negatively regulated by certain growth factors, including
bFGF. Cyclin D1 was induced in myoblasts by bFGF, and
stable expression of cyclin D1 inhibited C2C12 myoblast
differentiation. Cyclin D1 inhibited myoblast differentiation
by certain growth factors (161) and directly inhibited myogenic HLH proteins through an acidic region in cyclin D1’s
carboxy terminus (27). Forced expression of cyclin D1, but
not cyclins A, B or E, inhibited the ability of MyoD to transactivate muscle-specific genes and correlated with phosphorylation of MyoD. There are also pRB-independent mechanisms by which cyclin D1 inhibits growth factor-mediated
differentiation (162). Transfection of myoblasts with CKIs
p21Cip1 and p16Ink4a augmented muscle-specific gene expression in cells maintained in high concentrations of serum,
suggesting that an active cyclin-Cdk complex suppresses
MyoD function in proliferating cells (163). The direct binding
and inhibition of MyoD by Cdk4, together with the ability of
cyclin D1 to promote nuclear translocation of Cdk4, may
contribute to the inhibition of myocyte differentiation by
cyclin D1 (164). The possibility that the carboxyl termiunus
of cyclin D1, which was required for inhibition of MyoD (27),
may contribute to inhibition of differentiation through distinct mechanisms, including recruitment of histone acetylase-regulatory proteins (below), remains to be examined.
d. IGFs. As noted above, IGF-I has been classified as a
progression factor in the cell cycle. In density-arrested
BALB/c-3T3 cells made competent with PDGF treatment,
IGF-I with EGF induced G1 phase progression and initiation
of DNA synthesis. Subnanogram amounts of IGF-I with EGF
allow progression to the V-point but not S phase. Once cells
509
have progressed to the V-point within G1, IGF-I alone is
sufficient to promote progression into S-phase (165). Transcription of new mRNAs do not appear to be required for
IGF-I to function as a progression factor, and IGF-I is known
to stimulate posttranslational modifications of several proteins (166, 167).
IGF-I and IGF-II were isolated from human plasma as
peptides with insulin- and somatomedin-like properties
(168). Signaling by IGF-I requires a tetrameric type II IGF
receptor. The requirement for IGF and the IGF receptor in
normal growth was well illustrated by studies using targeted
disruption of the IGF-I receptor. These mice were growth
retarded, their size being 30% of wild-type control (169, 170).
The mouse embryo fibroblasts (MEFs) derived from these
animals also showed defective mitogenic capabilities, failing
to grow in medium supplemented with growth factors that
sustained the growth of wild-type cells (171). In the knockout
MEFs, all phases of the cell cycle were delayed (171, 172), and
this abnormality was reverted with reintroduction of the
wild-type receptor (171). The IGF-I receptor is also required
for EGF receptor function to exert its mitogenic effect (173).
IGF-I induces DNA synthesis in a broad array of cell types,
and intracellular signaling involves activation of Ras via a
Grb2-mSOS pathway. There is also a requirement for a docking protein IRS-1 in IGF signaling, which in turn regulates the
activity of SH2 domain-containing proteins such as PI3 kinase, Grb-2, Nck, and Syp (174, 175). The induction of cell
cycle progression by IGF-I treatment correlated with the
induction of cyclin D1 mRNA levels in T47D breast cancer
cells (176), in quiescent WI-38 cells (177), and MG63 osteosarcoma cells (178). The induction of cyclin D1 mRNA was,
at least in part, regulated at the level of transcription, and the
human cyclin D1 promoter linked to a luciferase reporter
gene was induced by IGF-I in cultured cells (179). Recent
studies suggested that IGF-I may also regulate cyclin D1
levels in vivo. During development, cyclin D1 is expressed in
proliferating cells of the developing CNS (179, 180) and retina
(181, 182). Protein-calorie malnutrition inhibits neonatal cerebellar growth and development, in part, through a reduction in IGF-I. Developmental delay induced by malnutrition
was associated with reduced cyclin D1 protein and CD1K
activity assessed using glutathione-S-transferase-pRB as a
synthetic substrate (179, 180). Nutritional replacement induced cyclin D1 protein levels and kinase activity in conjunction with a partial restoration of normal cerebellar development (179, 180).
As with PDGF, IGF also regulates the abundance of
p27Kip1. The induction of p27Kip1 protein levels that occurs
with growth factor withdrawal is likely mediated through
reduced ubiquitin-mediated degradation (98). Although the
reduction in p27 by interleukin-2 was blocked by rapamycin
(183), rapamycin did not block insulin-mediated inhibition of
p27Kip1 levels in Swiss 3T3 cells (184), suggesting a rapamycin-independent pathway is involved in insulin-mediated
DNA synthesis (184). Using a surrogate in vivo model of IGF-I
deficiency, nutritional deprivation-induced cerebellar developmental arrest was associated with a reduction in p27Kip1
levels from the same tissues in which CD1K activity was
decreased (180). These findings are consistent with the grow-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
510
PESTELL ET AL.
ing evidence from in vitro experiments that p21-related proteins may function to enhance pRB kinase activity (80).
e. EGF. EGF, like IGF, functions as a progression factor.
EGF is a potent mitogen for cells of ectodermal and mesodermal origin and is the prototype of a large superfamily of
ligands that signal through a family of related receptors. EGF
binding leads to receptor aggregation, autophosphorylation
and phosphorylation of intracellular substrates including
phospholipase C-g, MAP kinase, and the Ras GTPase-activating protein (GAP). Binding of EGF to the EGF receptor
(EGFR) can also induce heterodimerization with other members of the EGFR family, including c-ErbB-2 (185).
EGF binding to the EGFR induces expression of immediate
early genes such as c-jun, c-fos, and c-myc (186). In fibroblast cells
induced to proliferate by EGF, cyclin D1 levels were increased
(154). The induction of cyclin D1 protein was preceded by the
induction of cyclin D1 promoter activity in JEG-3 and CHO cells
(187). Dominant negative mutants of Ras, mitogen-activated
protein kinase (MAPK), and Ets inhibited EGF induction of the
cyclin D1 promoter, mapping a likely signal transduction pathway from mitogenic signals directly to the cell cycle-regulatory
apparatus (187). The EGF-related ligand TGFa in esophageal
and colon cancer cells (188) and activating mutants of c-ErbB-2
in MCF7 breast cancer cells (R. J. Lee, C. Albanese, G. Watanabe,
G. K. I. Haines, P. M. Siegel, W. J. Muller, M. C. Hung, and R.
G. Pestell, submitted) were also shown to regulate cyclin D1
abundance and promoter activity. Point mutation in the context
of the native cyclin D1 promoter demonstrated that the induction of cyclin D1 by c-ErbB-2 involved the E2F- and Sp1-binding
sites (R. J. Lee, C. Albanese, G. Watanabe, G. K. I. Haines, P. M.
Siegel, W. J. Muller, M. C. Hung, and R. G. Pestell, submitted),
and deletion of both the E2F and Sp1 sequences reduced TGFa
induction (188).
In PC12 cells, EGF stimulated cellular proliferation and
increased the levels of several cell cycle progression factors
including Cdk2, Cdk4, and cyclin B1 (190). p27Kip1 levels
were reduced by EGF in NIH3T3 cells, and dominant negative mutants of Ras blocked the EGF-induced down-regulation of p27Kip1, consistent with dual roles of Ras in early G1
and late G1/S (191). As with PDGF, EGF-mediated induction
of cyclin D1 and inhibition of p27Kip1 abundance appears to
be a common finding in several different cell types.
f. Nerve growth factor (NGF). The differentiation and survival of CNS and peripheral nervous system (PNS) neurons
is influenced by neurotrophic factors including NGF, brainderived neurotrophic factor (BDNF), ciliary neurotrophic
factors, and the fibroblast growth factors. One of the better
characterized neurotrophins, NGF, is required for the differentiation and survival of sympathetic and sensory neurons of the PNS. NGF binds to two transmembrane proteins,
p140trk and p75, on the cell surface (192). PC12 cells, which
are derived from a neural crest-derived pheochromocytoma
cell line, respond to NGF by withdrawing from the cell cycle,
extending neurites and acquiring features of sympathetic
neurons. NGF induces a sustained activation of the MAPK
pathway and induces a pp90Rsk kinase (193). Activation of
the MAPK pathway by an activating Raf mutant in NIH3T3
cells overexpressing the TrkA receptor is sufficient to induce
p21Cip1 and growth arrest (194).
The response of cell cycle-regulatory proteins to NGF sug-
Vol. 20, No. 4
gests a concurrent induction of G1 cyclins, and CKIs may
participate in coordinating the differentiation signal. NGF
induces expression of cyclin D1 and the CKI p21Cip1 (195–
197). In association with these increases in cyclin D1 and
p21Cip1, the protein levels and associated activities of Cdk4,
Cdk6, and cdc2 decreased. The induction of the cyclin D1 and
p21Cip1 genes by NGF requires DNA sequences in their respective promoters that include Sp1-binding sites (198).
The induction of cyclin D1 is not sufficient for the induction of neurite outgrowth in PC12 cells, however, raising the
possibility that the induction of cyclin D1 by NGF subserves
a distinct function (196). Because the addition of Cdk chemical inhibitors to undifferentiated PC12 cells promoted cell
death, it was suggested that cyclin D1 may convey a cell
survival function (199). During neuronal apoptosis, however, CD1K activity was stimulated and cyclin D1 was required for the induction of apoptosis in R970B cells (200). The
induction of apoptosis by cyclin D1 did not require p53 and
was inhibited by Bcl-2 and the 21-kDa E1B protein, suggesting cyclin D1 inhibited cell survival (200). Expression of
cyclin D1 in the HC11 breast cell line (201) or NIH3T3 cells
(200) also induced apoptosis, and the induction of apoptosis
in rat fibroblasts was observed with cyclin D1 but not with
cyclin E (202). It remains possible, therefore, that cyclin D1
may either promote apoptosis or function as a sensor of
impending cell death. It has been suggested that the induction of apoptosis by cyclin D1, particularly after ionizing
radiation, represents a G1/S checkpoint function of cyclin D1
(30, 203) (for a review see Ref. 4). Under these circumstances,
cyclin D1 is actually required for the cell cycle arrest, and a
similar function was observed for cyclin D1 in p53-mediated
cell cycle arrest (204). In part, this cell cycle-arrest function
of cyclin D1 may be through its ability to bind PCNA and
sequester its activity in the nucleus.
Recent studies have suggested a role for Cdk5 in neurite
outgrowth, and overexpression of a Cdk5 dominant negative
mutant blocked neurite extension in cultured cortical neuron
cultures (205). The heterodimeric partners for Cdk5, p35, and
the related p39 are required for normal neurite extension in
a rat hippocampal cell line (58). The role of the cyclin/Cdk
complexes in NGF signaling remains to be fully explored.
The abundance of other cell cycle-control proteins are altered
during NGF treatment of PC12 cells, although their role in
mediating the differentiated phenotype is not understood. One
of the earlier changes induced in PC12 cells treated with NGF
is a decrease in cyclin F, raising the possibility that cyclin F is
also involved in NGF-mediated cell cycle events during the
differentiation of PC12EY cells (206). In addition the DNA binding activity of the E2F/DP proteins, which bind DNA enhancer
sequences in a number of different genes involved in promoting
cell cycle progression, was reduced with NGF treatment (195).
2. Serine/threonine kinase receptors.
a. TGFb. The TGFb family of cytokines regulates diverse
functions including differentiation and inhibition of cell
growth (207). In addition, TGFb can induce cellular proliferation in certain cell types, such as subconjunctival and
adult lung fibroblasts (208, 209). The proliferative effects are
likely in part related to release of paracrine growth factors
such as connective tissue growth factor (210). TGFb inhibits
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
cellular growth in most epithelial cells and can act at both
early and late points during the prereplicative G1 period
(211–213). The diverse effects of TGFb are, in part, regulated
by the cell type, the state of cellular differentiation, local
paracrine context, and the relative abundance of the TGFb
receptors. The signaling pathway induced by TGFb is dependent upon binding of ligand to the TGFb type II receptor,
which forms a heteromultimeric transphosphorylated complex with the TGFb1 receptor.
Studies of pathways activated by TGFb indicate important
roles for members of a multigene family related to Drosophila
Mad (mothers against decapentaplegia) in intracellular signaling. In addition the CKIs appear to play an important role
in the cytostatic phenotype induced by TGFb. In the developing Drosophila eye (214), signaling mediated by the TGFbrelated gene decapentaplegia (dpp) was required for the
synchronization of the cell cycle. dpp May affect cell cycle
synchronization by promoting cell cycle progression. This
synchronization is critical for the precise assembly of the
Drosophila eye. More than six mammalian proteins related to
Drosophila Mad, Smads, have been cloned and are implicated
in transducing TGFb signaling to the nucleus, with Smad3
and Smad4 functioning as DNA sequence-specific binding
proteins (215–219).
The effect of TGFb on components of the cell cycle-regulatory apparatus are quite complex (211). TGFb functions in
part through both transcriptional effects and posttranscriptional and posttranslational mechanisms. For example, the
inhibitory effect of TGFb on CCL64 cells late in G1 occurred
in the presence of RNA synthesis inhibitors (220). Unifying
themes are appearing, however, indicating a critical role for
the CKIs (p21Cip1, p27Cip1 and p15Ink4b) in which the transcriptional induction and subnuclear localization appear to
mediate their cell cycle inhibitor effects. In addition, TGFb
has been shown to inhibit expression of Cdk2, cyclin D2, and
cyclin A and to reduce pRB phosphorylation (221). TGFb
inhibits growth in fibroblasts and Mv1Lu cells in association
with a delayed reduction in Cdk4 abundance (222) while in
several cell types it appears that the earliest and critical steps
in the cytostatic function of TGFb is the induction of the
Cdk4/6 inhibitor p15(Ink4b/MTS2). TGFb stabilizes p15Ink4b
protein, increases p15Ink4b-Cdk4 complexes, and inhibits cyclin D1/Cdk4 association in human mammary epithelial
cells (223). The recent studies from Massague’s laboratory
(92) have been particularly insightful in understanding TGFb
signaling to the CKIs with their recent finding that the subcellular locations of p15Ink4b and p27Kip1 coordinate their
inhibitory interactions with Cdk4 and Cdk2. In lung epithelial cells, treatment with TGFb led to an induction of p15Ink4b
bound to cyclin D1/Cdk4 and reduced binding of cyclin
D1/Cdk4 by p27Kip1. The displacement of p27Kip1 led to its
binding to cyclin E/Cdk2, which forms an inhibitory complex. These studies suggested the subcellular distribution,
and the proteins to which p27Kip1 was bound may be critical
in mediating the cytostatic effect of TGFb (92) (Fig. 5). In cells
lacking p15Ink4b, however, TGFb is still capable of arresting
cells in G1 through a reduction in cyclin D1 and an induction
of p21Cip1 and p27Kip1 (224).
TGFb can also inhibit Cdk4 and Cdk6 activities by increasing their level of tyrosine phosphorylation. The induc-
511
FIG. 5. A, TGFb has been shown to induce p21Cip1, p27Kip1, and p15
Ink4b
in different cell types. The induction of p21Cip1 is p53 independent, as with the induction of p21Cip1 by BRCA1. The role of BRCA1
in TGFb induction of p21Cip1 is not known. It is known that cyclin
D1/Cdk activity is reduced in TGFb-treated cells and that p21Cip1 can
inhibit cyclin D1. B, The addition of TGFb alters the associations of
p27Kip1 from cyclin D1/Cdk4 to a cyclin E/Cdk2 complex, the activity
of which it inhibits. p15Ink4b replaces p27Kip1 bound to cyclin D1/Cdk4,
inhibiting its activity. In this model the amount of p27Kip1 may not
change in the TGFb-treated cell.
tion of Cdk4/6 phosphorylation is the result of reduced
dephosphorylation through inhibiting expression of the Cdk
tyrosine phosphatase Cdc25A (225). Repression of Cdc25A
expression and the induction of p15Ink4B are independent
effects that together mediate the inhibition of Cdk4/6 activity
by TGFb (225). In primary fibroblasts, the growth inhibition
mediated by TGFb required the pRB (226). Recent studies
have suggested an important role for histone deacetylase in
the transcriptional repression mediated by pRB (below), and
the pocket proteins, pRb, p107, and p130, each appear to be
capable of binding HDAC (227). In transgenic mice overexpressing TGFb in the liver, which have impaired proliferative responses to in vivo stimuli, HDAC binding to p130 is
specifically enhanced (B. Bouzahzah, M. Fu, A. Iaavarone, E.
Bottinger, V. Factor, S. Thorgeirsson, and R. G. Pestell, submitted). It will be of interest to determine whether TGFb
modulates its cytostatic function in part through HDAC
binding to pocket proteins. Together, however, these studies
suggest that important cell type-specific differences determine both the effect of TGFb on cellular phenotype and the
effect on specific cell cycle-regulatory protein abundance and
phosphorylation.
b. Activins. Activins are members of the TGFb family encoded by two closely related genes, activin-bA and activinbB. Activins inhibit cellular proliferation and induce differ-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
512
PESTELL ET AL.
entiation of mesoderm, erythroid precursors, and other cell
types (229 –231). Activin also inhibited basal and stimulated
proliferation in prostate cancer cell lines (232). In plasmacytic
cells, the induction of cell cycle arrest by activin A was
associated with an induction of p21Cip1, but not the other
CKIs (p27Kip1, p16Ink4a, or p15Ink4b), and a suppression of
cyclin D2 (233). Because pRB phosphorylation was reduced
in association with the induction of p21Cip1 in activin Atreated cells, the cell cycle arrest induced by activin was
attributed to the induction of 21Cip1 expression.
B. Seven-transmembrane receptors
1. Rhodopsin family.
a. Angiotensin II (AII). The octapeptide AII binds to specific
high-affinity receptors present in the adrenal cortex, liver
epithelial cells, and in vascular smooth muscle cells where it
elicits a vast array of biological effects. AII increases DNA
synthesis, cell proliferation, and steroidogenesis in cultured
adrenal cortical cells, both in cells derived from the adrenal
fasciculata and glomerulosa cell layer (234, 235). AII functions as a growth factor in cardiac fibroblasts, myocytes,
vascular smooth muscle cells, and adrenal cells (236 –238).
Many of the known biological actions of AII, including enhanced DNA synthesis, are mediated by stimulation of the
AT1 receptor (234, 235).
The AT1 receptor is a member of the G protein-coupled
seven-transmembrane spanning receptor family (239, 240).
Binding of AII to the AT1 receptor activates phospholipase C,
which initiates a rapid release of inositol triphosphate and
diacylglycerol from phosphatidylinositol 4,5-bisphosphate,
causing intracellular calcium release (241). The intracellular
transmission of signaling by Ca12 and activation of cytosolic
phospholipases involves, in part, sequential activation of Ras
(242–244) and thereby protein kinases, including MAPK (240,
245). Previous studies showed that AII can stimulate phosphorylation of several intracellular signaling protein kinases
at tyrosine residues including the ERKs in vascular cells (246,
247) and the related stress-activated protein kinases [SAPKs
or Jun N-terminal kinases (JNKs)] in hepatic cells (248). In
addition, AII stimulates tyrosine phosphorylation of p44/
p56SHC (246), the Jak family proteins Jak2 and Tyk2 (249), and
focal adhesion kinase (p125FAK) in vascular smooth muscle
cells (246).
In the human adrenal cell line H295R, AII (1026 m) stimulated G1 phase progression within 12 h, with a maximal
effect after 72 h (250). This action was preceded by the induction of cyclin D1 mRNA, the presence of nuclear cyclin
D1 protein, and CD1K activity. Acting through the AT1 receptor, AII induced cyclin D1 promoter activity 4-fold via an
enhancer sequence at 2954 bp. c-Fos and c-Jun bound the
cyclin D1 2954 enhancer sequence, and the abundance of
c-Fos within this complex was increased by AII treatment
(250). AII induced ERK activity 7-fold, and dominant negative mutants of either Ras or ERK reduced AII-stimulated
cyclin D1 promoter activity. These findings suggest AII may
stimulate mitogenesis by increasing CD1K activity through a
Ras/ERK/AP-1 pathway.
b. Parathyroid hormone (PTH). Osteoblasts are the major
direct cellular target of PTH action in bone. The high-affinity
Vol. 20, No. 4
receptors for PTH generate a variety of intracellular signaling
pathways, including changes in intracellular cAMP, inositol
triphosphate, diacyl glycerol, Ca12, membrane potential, and
pH. The effect of PTH on osteoblast proliferation and differentiation is complex, and PTH may either inhibit or induce
cellular proliferation depending upon the cell system examined and upon whether the exposure to PTH is tonic or
intermittent. Acute exposure to high concentrations of PTH
leads to an inhibition of many osteoblastic functions. Continuous exposure to high concentration of PTH in vivo results
in a progressive bone loss and osteopenia, although osteoblasts may increase in number. PTH is known to inhibit
proliferation of the UMR-106 osteoblast cell lines and reduce
the proportion of cells entering S-phase. Associated with
these changes, PTH increased p27Kip1, but not p21Cip1 levels.
This effect was mimicked by 8-bromo-cAMP, but not by
phorbol 12-myristate 13-acetate. The protein kinase A inhibitor KT5720 abolished the PTH induction of p27Kip1. PTH
increased Cdk2-associated p27Kip1 without affecting the levels of Cdk2 (251). Extracellular calcium inhibits parathyroid
cell proliferation. In a rat epithelial parathyroid cell line, PT-r,
cyclin D1 mRNA levels oscillate during the cell cycle, and
increasing amounts of calcium in the incubation medium
reduced the expression of rat cyclins D1 and D2 (252).
c. TSH. TSH is a member of a dimeric family of glycoprotein hormones that contain a common a- and a unique
b-subunit. The binding of TSH to its cognate G proteincoupled receptor (Gs) initiates a cascade of intracellular signaling, including the induction of cAMP, and in the human
the activation of phospholipase C (253). The cAMP pathway
induces DNA synthesis with kinetics comparable to the effect
of TSH and is associated with the induction of immediate
early genes and PCNA (254). In the rat thyroid cancer cell line
FRTL5, TSH induces cellular proliferation and cell cycle progression. A FRTL5 cell line containing a dominant negative
mutant of CREB (cAMP reponse element binding protein)
had reduced induction of DNA synthesis in response to TSH,
providing supportive evidence that the cAMP/CREB pathway was linked to DNA synthesis in thyroid cells (255). The
proliferative effects of TSH are associated with an enhanced
rate of G1 phase progression and induction of cyclin D1 and
cyclin E (256). The phosphatase inhibitor okadaic acid, which
inhibits protein phosphatase 2A (PP2A) and protein phosphatase 1, stimulates the TSH-induced G1-S phase transition
in thyroid cells (257). In dog thyrocytes, microinjection of
human GST-p16 inhibited BrdU incorporation induced by
TSH (18). Cyclin D3 abundance increased in the nucleus of
TSH-treated dog thyrocytes, and microinjection of immunoneutralizing cyclin D3 antibodies blocked TSH-induced
BrdU uptake, suggesting a specific role for cyclin D3 in G1
phase entry induced by TSH (258).
d. FSH. The binding of FSH to its seven membrane-spanning receptor leads to induction of cAMP within the cell
through heterodimeric G protein coupling to adenyl cyclase
(259). FSH induces ovarian folliculogenesis and male spermatogenesis, and the mitogenic effect of FSH is, in part,
induced through cAMP (259, 260). Many actions of FSH can
be mimicked by the addition of cAMP to cultured ovarian
cells. Cyclin D2 was found in FSH-responsive Sertoli cells
and spermatagonia; furthermore, testicular germ cell tumors
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
express very high levels of cyclin D2 (261). cAMP treatment
of granulosa cells induced cyclin D2 mRNA with similar
kinetics to the induction by FSH (261, 262). Serum addition
or induction of the protein kinase C (PKC) pathway by mitogens did not induce cyclin D2 mRNA levels, suggesting
activation of the cAMP pathway by FSH was critical for the
induction of cyclin D2 mRNA. Homozygous deletion of the
cyclin D2 gene resulted in female mice with hypoplastic
granulosa cells, unresponsive to FSH, suggesting a critical
role for cyclin D2 in FSH-induced cellular proliferation in vivo
(261). The male cyclin D2 KO mice displayed hypoplastic
testis, with normal differentiation and normal serum testosterone levels, suggesting an important role for cyclin D2 in
cellular proliferation required for the formation of a critical
mass of tissue during development.
C. Steroid hormones
1. Estrogens. Steroid hormones mediate diverse effects on
cellular proliferation in association with modulating activity
of the cyclins and the CKIs. The proliferative effects of estrogen on responsive tissues, including breast and uterus,
have been well documented (for reviews see Refs. 263–265).
The use of animals homozygously deleted of the ER-a gene
(ERa KO mice) confirmed the requirement of the ER for
several normal proliferative processes including normal
mammary gland ductal growth (266, 267), angiogenesis
(268), and spermatogenesis (269).
A primary mechanism of estrogen action is the ligand
dependent steroid hormone receptor activation through specific response elements of target genes (270, 271). In addition,
estradiol regulates the Ras-Raf-MAPK pathway with kinetics
similar to those of polypeptide growth factors operating
through membrane tyrosine kinase receptors (272). Peptide
growth factors also modulate ER activity independently of
ligand (273), and the ER is capable of interacting with a
number of other transcription factors to coordinate expression of downstream target genes, including members of the
AP-1 family (274) and Sp1. In part, the coordinate regulation
of these downstream transcription factors is regulated by
interaction with high molecular weight co-integrator
proteins, including SRC-1, RIP-140, SNF2b-BRG1 (275),
TASF(II)130 (276), and TIFI (277) (reviewed in Ref. 278).
Recent studies have focused on the molecular mechanisms
by which estrogens induce components of the cell cycleregulatory apparatus to induce cellular proliferation. Estrogens stimulate cell cycle progression early in G1 phase in
cultured breast epithelial cells (279 –281). Cyclin D1 expression is reduced by antiestrogen treatment in T47-D cells (176).
The induction of cellular proliferation by estrogen in breast
cancer cell lines was found to correlate with increased expression of cyclin D1 protein levels and CD1K activity in
MCF-7 and T-47D cells (282–284). In the experiments conducted by Altucci, the MCF-7 cells were released from cell
cycle arrest by 24 – 48 h of treatment with the hydroxymethylglutyryl-coenzyme A reductase inhibitor, simvastatin, before the addition of estrogen. Under these experimental conditions, cyclin D1 levels were induced rapidly between 1–3
h, and the induction of cyclin D1 mRNA was blocked by
actinomycin D, suggesting a role for RNA polymerase II. The
513
cyclin D1 promoter was also induced by estrogens, indicating the induction by estrogen may be a direct transcriptional
event (283).
The recent series of studies demonstrating the activation
of the unliganded ER by EGF through the Ras-ERK pathway
suggest additional mechanisms by which growth factors and
estrogen may regulate cyclin D1 expression (285). The addition of estrogen to lovastatin-treated cells induced CD1K
activity. The induction of DNA synthesis by estrogens was
inhibited by the microinjection of cyclin D1-neutralizing antibodies but not by dominant negative mutants of the MAPK
kinase pathway (18). These results suggest that the induction
of cyclin D1 is necessary whereas the induction of ERK activity that occurs in response to estrogens in MCF7 cells is not
directly responsible for the induction of S phase entry (18).
The CKIs are also important in estrogen-induced mitogenesis in breast cancer cell lines (Fig. 6B). Overexpression of
p16Ink4a, for example, blocked the estrogen-induced S phase
entry in MCF7 cells (18), indicating a critical role for the CKIs
in estrogen-induced mitogenesis. The mechanisms by which
estrogen regulates CKI function can be mechanistically considered as three distinct but functionally interrelated effects.
First, estrogen treatment induces alterations in the subcellular localization of the CKI. Estrogens induce cyclin E/Cdk2
activity in association with an alteration in the relative distribution of p21Cip1 from an inhibitory cyclin E/Cdk2 complex to the cyclin D1/Cdk4 complex (286). Because p21Cip1
can inhibit cyclin E/Cdk2 activity, but may, at specific stoichiometric ratios, foster cyclin D1/Cdk4 activity, the net
effect of the relocation of p21Cip1 is to promote cell cycle
progression and cellular proliferation (286). Second, estrogen
alters the nature of the multimeric complexes formed between the cyclin/Cdk complexes. In studies by Prall et al.
(287), estrogens reduced the amount of the CKIs p21Cip1 and
p27Kip1 protein bound to cyclin E/Cdk2 (Fig. 6B). Third,
estrogen treatment results in the formation of higher molecular weight complexes of cyclin E/Cdk2, which lack p21Cip1
and p27Kip1. These high molecular weight complexes contained increased Cdk2 phosphorylation at Thr 160. In part,
therefore, the estrogen-induced activation of cyclin E/Cdk2
appeared to involve both a reduction in the associated CKIs
and increased CAK activity. The use of gel filtration chromatography was critical to demonstrating the alterations in
the cyclin/Cdk/CKI multimeric complexes. The nature of
these high molecular weight complexes is currently unknown; however, it is possible that these complexes include
ER coactivator proteins, such as SRC-1 or p300.
Of interest in these studies of estrogen action is the common finding of combined induction of cyclin D1/Cdk4 kinase activity and cyclin E/Cdk2 activity by estrogens. Myc
expression is also induced by estrogens, and forced overexpression of Myc in MCF7 cells induces changes in cyclin
E/Cdk2 activity similar to those induced by estrogen treatment (288). Thus, overexpression of Myc, cyclin D1, or estrogen treatment each result in decreased association of
p21Cip1 with cyclin E/Cdk2 and result in the formation of
a high molecular weight multiprotein complex with high
levels of histone H1 kinase activity (288).
Recent studies using mammary gland transplants from
ERa knockout mice suggest that stromal cells play a role in
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
514
PESTELL ET AL.
Vol. 20, No. 4
FIG. 6. Mechanisms by which estrogen regulates cell cycle proteins and cellular proliferation. A, Induction of cyclin D1 gene expression.
Estrogen induces expression of cyclin D1. Cyclin D1 binds Cdk4, and this complex phosphorylates nuclear pRB protein, leading to cellular
proliferation. p16Ink4a inhibits estrogen-induced mitogenesis, further supporting a model in which estrogen-regulated cellular proliferation
requires cyclin/Cdk. B, Estrogen regulates the subcellular distribution of p27Kip1. p21Cip1 and p27Kip1 associate with cyclin D1/Cdk4 complexes.
Estrogen treatment alters p27Kip1 and p21Cip1 association with Cdk2/cyclin E. p21Cip association with cyclin E/Cdk2 was reduced while
association with cyclin D1/Cdk4 was increased (286). Cdk2 phosphorylation at Thr-160 was increased by estrogen treatment, associated with
an increase in Cdk2 activity. C, Cyclin D1 binds to the ER. The binding of cyclin D1 to the ER promoted ER-dependent enhancer activity through
an estrogen response element (ERE) (114, 115). This induction occurs independently of the Cdk-binding properties of cyclin D1, perhaps involving
recruitment of coactivator proteins such as p300 or associated histone acetylase activity. D, Cyclin A/Cdk2 phosphorylates the ER. Phosphorylation of the ER by cyclin A/Cdk2 directly alters its transcriptional activity (292).
the mitogenic effects of estrogens (289). Mammary tissue
from ER-deficient mice and wild-type mice were used to
produce tissue recombinants that were grown as subrenal
capsule grafts in intact female nude mice. In these experiments ductal growth in the ovariectomized hosts treated
with estrogen required stromal ER but not epithelial ER
(289).
Cyclin D1 binds directly to the ER and regulates estrogendependent enhancer activity (114, 115). The transactivation
function of the ER, when fused to a GAL-4 DNA-binding
domain, was enhanced by overexpression of cyclin D1 (Fig.
6C). This activity was induced further by the addition of
estrogens, an effect that was mediated through the EF domain of the ER (114, 115). Cyclin D1 activated the ER-mediated transcription in the absence of ligand, and the induction occurred independently of the Cdk- or pRB-binding
domains of cyclin D1 (114, 115). Because ER status is often
positive in postmenopausal women, it has been proposed
that the overexpression of cyclin D1, frequently seen in ERpositive tumors, may function to promote ER activation of
target genes in the presence of low estrogen levels. In analysis
of the molecular mechanisms by which cyclin D1 enhances
ER activity, cyclin D1 was found to bind the ER coactivator
SRC-1 through a carboxy-terminal LLXXXL motif, with de-
letion of this region reducing ER activation by 80% (290).
Because the LXXL motif of SRC-1, which was required for
cyclin D1 binding (290), is a conserved motif among several
coactivators (291), these studies imply cyclin D1 may interact
with other coactivator proteins. These studies demonstrated
that cyclin D1 may stimulate estrogen enhancer activity and
provided further evidence for a model in which cyclin D1
interacts with proteins other than the Cdks to mediate hormonal effects on target gene sequences.
Cyclin A overexpression also enhanced ER activity, and
ER is phosphorylated between amino acids 82 and 121 in vitro
by cyclin A, an effect that was inhibited by p27Kip1 overexpression (292) (Fig. 6D). Together, these results suggest that
the regulation of the Cdks by estrogens is under complex
feedback loops in which the transcriptional targets also serve
as substrates. Most of the studies described above have focused on the ERa gene, although recent studies have shown
that the ERb gene mRNA abundance appears to be regulated
by the ERa gene (293) and that the ERa and ERb genes can
differentially regulate AP-1 activity (294).
The effects of estrogens in other cell types may also be
under equally complex regulatory pathways. In the rat
uterus, estrogen induces endometrial epithelial cell proliferation (295). The administration of 17b-estradiol (E2) to
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
ovariectomized rats induced Cdk4, -5, and -6, but not Cdk2
activation in the uterus, and increased expression of cyclin
D1 and D3, cyclin A, and cyclin E mRNAs (295). p27Kip1 was
also induced by E2 in the uterus (295). It will be of interest
to determine whether the mechanisms regulating the effects
of specific synthetic estrogens differ in different tissues.
2. Progestigens. The physiological functions of progesterone
are mediated by two naturally occurring receptor isoforms,
PRA and PRB, derived from a common gene. The progesterone receptor (PR), as with other nuclear receptors, contains
DNA-binding, ligand-binding, and transactivation regions.
Evidence for a normal role for progesterone in promoting
mammary epithelial cell growth during puberty and pregnancy was supported by the augmentation of mammary
gland growth by the administration of progesterone (296,
297), the inhibition of mammary gland tumor growth by
antiprogestins (298), the positive correlation between proliferative potential and PR abundance, and the finding that PR
mRNA was concentrated at the end bud cells and in the
undifferentiated progenitors of the mammary gland duct
wall (299). Transgenic mice homozygously deleted of both
PR isoforms provided further support for progesterone’s
critical role in mammary gland proliferation (300). In the PR
KO mice, mammary gland development was severely limited with absence of end buds and failure of lobuloalveolar
development. The animals were unable to ovulate and displayed uterine hyperplasia consistent with the loss of the
progesterone decidual response.
In breast cancer cell lines, progestigens both stimulate and
inhibit cell cycle progression (176, 265, 301, 302). The initial
response is acceleration of cells from G1 to S and then, upon
completion of the cell cycle, the progestin-treated cells arrest
in G1 (176, 265, 301, 302). The transient induction of G1 phase
by progestigens is accompanied by an induction of cyclin D1.
However, the inhibition of cell cycle progression by antiprogestigen treatment in T47D cells is not accompanied by
changes in cyclin D1 expression (176). The reduction in CD1K
activity that occurs with antiprogestigens, such as RU486 or
ORG 31710, is associated with an induction of p21Cip1 mRNA
and protein within the cyclin D and cyclin E kinase complexes. RU486 has potent antiglucocorticoid activity whereas
ORG 31710 is thought to be more selective for its antiprogestigen activity. Overexpression of cyclin D1 markedly reduced the antiproliferative effect of the antiprogestigens
(303). Progesterone enhances the induction of cyclin D1 by
estrogen in the murine mammary gland, and this induction
was abolished in animals homozygously deleted of the PR
(304), indicating the effect was likely mediated through the
PR. The induction of p21Cip1 may also contribute to the differentiation and apoptosis-inducing properties of progestins
that are observed after prolonged treatment, as p21Cip1 overexpression is capable of inducing differentiation and apoptosis in other cell types. c-Myc mRNA levels rose before the
induction of cyclin D1 mRNA levels, raising the possibility
that c-myc may be a positive regulator of cyclin D1 expression
in the presence of progestin (176).
3. Retinoic acid. The highly pleiotropic effects of retinoic acids
in regulating proliferative and differentiation pathways in
515
mammalian cells (305) result in part through combinatorial
interactions of their receptors (306, 307). The three retinoic
acid receptors (RARa, -b, and -g) and retinoid X receptors
(RXRa, -b, and -g) and their isoforms exhibit distinct patterns
of expression. All-trans- and 9-cis- retinoic acids are among
the physiological retinoids. RARs and RXRs act as ligandinducible transcription factors, with the homo- and heterodimers binding distinct DNA sequences. Retinoids had
been shown to affect cellular proliferation at many different
levels, and the predominant effect is to inhibit cellular proliferation and induce differentiation (305, 308). The expression of receptors that bind growth factors was shown to be
directly inhibited by retinoids (309). The retinoids have also
been shown to directly affect expression of cell cycle-regulatory genes. The p21Cip1 gene, for example, is a retinoic
acid-responsive target gene, and an RA response element in
the promoter is required to confer retinoic acid induction
through RAR/RXR heterodimers (310). The induction of
monocytic differentiation by RA in U937 cells correlated with
the transcriptional activation of the p21Cip1 gene and suggests a role for this cyclin/Cdk complex inhibitor in facilitating this differentiation pathway. The RA-mediated inhibition of cyclin D1 protein in bronchial epithelial cells is
regulated at the posttranslational level, likely through increased ubiquitin-dependent proteasome degradation. In
these studies, calpain inhibitor I and lactacystin each prevented the decrease in cyclin D1 protein expression in the
presence of RA treatment (311). Free E2F-1, which is induced
by phosphorylation of pRB, inhibited RARa transactivation
function but did not inhibit glucocorticoid receptor (GR) or
thyroid hormone receptor (TR) transactivation (312).
The RARs and RXRs also act as ligand-dependent transrepressors of AP-1 (activator protein-1) (c-Jun/c-Fos) activity, and reciprocally AP-1 can inhibit transactivation by
RARs and RXR (313, 314). Synthetic retinoids were identified
that selectively enhanced the activation function of RARb
and efficiently inhibited AP-1 activity. These activities were
associated with an inhibition of anchorage-independent
growth (315). These studies suggested that retinoid-induced
growth inhibition may be related to AP-1 transrepression.
The identification of high molecular weight coactivator proteins, CBP and p300, required for both nuclear receptor activation and the inhibition of AP-1 activity (316), raised the
possibility that limiting abundance of coregulatory proteins
may dictate the genomic response to the addition of a receptor ligand (reviewed in Ref. 317). Linking activity of these
rate-limiting coregulatory proteins to the regulation of genes
that encode rate-limiting steps in cellular proliferation may
provide important insights into the antiproliferative effects
of retinoids. In this context it will be of considerable interest
to determine whether the RXR receptors, like the ER, are
capable of binding to cyclin D1 and directly modulating cell
cycle function.
4. Glucocorticoids. The glucocorticoid receptor is a member of
the intracellular receptor superfamily of transcriptional regulatory proteins and contains separable transactivation, hormone-binding, and DNA-binding domains (270, 318). The
activity of the GR is regulated in a cell cycle-dependent
manner and glucocorticoids directly affect cellular prolifer-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
516
PESTELL ET AL.
ation and cell cycle progression. The action of glucocorticoids
is cell cycle dependent. The inability of glucocorticoids to
induce gene expression during the G2 phase of the mammalian cell cycle has long been known (319). In cultured rat
hepatoma cells, glucocorticoids induce tyrosine amino transferase (TAT) activity in late G1 and S phases but not during
G2/M and early G1 phases (319). Glucocorticoid induction of
several other genes, including the metallothionein-1 gene
(320), were subsequently shown to be inhibited during the G2
phase of the cell cycle (321). It was observed that G2-arrested
cells contained a modified form of the GR with altered chromatographic properties (322), and it was observed that GRs
that translocate to the nucleus in G2 in response to dexamethasone treatment were inefficiently retained and redistributed rapidly to the cytoplasm. The inefficient nuclear
retention of the GR in G2-arrested cells was associated with
altered phosphorylation of the GR, and it was proposed that
dephosphorylation of the GR may be important in its nuclear
retention (320).
The amino terminus contains several sites that undergo
ligand-dependent (323) and cell cycle-dependent phosphorylation, with basal GR phosphorylation being higher in
G2/M than in S phase (320, 324). Selective mutations of single
or multiple phosphorylation sites of the mouse (325) or the
human GR (326) only reduced the transcriptional activity
modestly. The phosphorylation status of the GR, however,
may co-determine its subcellular location (327) and interaction with other transcription factors or coactivators. Four
major sites within the rat GR are phosphorylated in the
presence of hormone: S224 and S232 are phosphorylated by
the Cdks, whereas the constitutively phosphorylated residues, T171 and S246, are phosphorylated by members of the
MAPK family (112) (Fig. 7). Cyclin E/Cdk2, cyclin A/Cdk2,
and cyclin B/cdc2 were each capable of phosphorylating the
GR in vitro. The transcriptional activity of the GR was reduced in yeast strains deficient in particular G1 (CLN) and G2
(CLB) genes, whereas strains of yeast bearing deletions of
MAPK genes, FUS3 and KSS1, enhanced GR transactivation
function. Together these studies suggest that transactivation
function of the GR may be enhanced by several cell cycleregulatory kinases.
Glucocorticoids have been shown to inhibit cellular proliferation in a variety of cell types, with an inhibition occurring in mid-G1 phase. Glucocorticoid inhibition of L929 cell
proliferation was associated with an induction of p21Cip1
(328) and inhibition of alveolar epithelial cells entry into S
phase by specifically altering the activation of cyclin E/Cdk2
complexes through induction of the Cdk inhibitor p21Cip1
(329). Ectopic expression and activation of glucocorticoid
receptor in human osteosarcoma cell lines U2OS and SAOS2,
which lack endogenous receptors, result in a G1 cell cycle
arrest. GR activation in U2OS cells represses expression of
the Cdks, Cdk4 and Cdk6, as well as their regulatory partner,
cyclin D3, leading to hypophosphorylation of the retinoblastoma protein (Rb) (330). In pRB-deficient cells, the GR induction of cell cycle arrest is associated with the induction of
the expression of the Cdks, p21Cip1 and p27Kip1. The aminoterminal activation domain of the GR was required for
growth arrest in SAOS-2 cells consistent with the role of the
amino terminus in activation of gene transcription of the
Vol. 20, No. 4
FIG. 7. Mechanisms by which glucocorticoids regulate the cell cycle.
A, Functional map of the glucocorticoid receptor (GR) deduced from
analysis of the human, mouse, and rat receptors. The domains identified by partial proteolysis (amino-terminal, DNA-binding, and hormone-binding domains) are indicated. The sites of the GR affected by
phosphorylation during cell cycle transition are indicated by an asterisk. The sites in the rat GR that are phosphorylated by cyclin
A-Cdk2, cyclin E/Cdk2, and cyclin B/cdc2 are serines 224 and 232. The
MAPK, ERK, phosphorylates threonine 171 and serine 246 (112). B,
GR-mediated cell cycle arrest involves cell type-dependent mechanisms including 1) repression of the G1 cyclins and Cdks; 2) enhanced
expression of the CKIs (330); and 3) perhaps through induction of
IKBa and the reduction in cytoplasmic NFkB activity, reduced G1
cyclin kinase activity.
CKIs. In addition, the GR affects a number of other target
genes and transcription factors, which in turn may mediate
a component of the cell cycle-regulatory effects. For example,
the GR induces IKBa synthesis (331, 332), which binds NFkB, keeping it in its inactive form in the cytoplasm, and may
thereby regulate CKI abundance indirectly.
5. Vitamin D. In addition to a role in regulating calcium
homeostasis, immune responses, and cellular differentiation,
vitamin D and its metabolites play important roles in the
regulation of cellular proliferation and differentiation of normal and malignant cells (reviewed in Ref. 333). 1a,25(OH)2D3
generates biological responses through genomic and nongenomic pathways, and the genomic responses involve
homo- or heterodimerization with other nuclear receptors
including members of the RXR and T3 receptor family. The
antiproliferative effects of 1a,25(OH)2D3 and several vitamin
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
D analogs have been documented for human, mouse, or rat
breast cancer lines (334 –336) and implantable breast tumor
models (337–339), keratinocytes (340, 341), and prostate cancer cell lines (342).
The inhibition of human and murine keratinocyte proliferation by vitamin D3 was associated with a decreased number of EGFRs, a rapid decrease in c-myc expression (343), an
induction of TGFb (344), and a reduction in pRB phosphorylation (345).
The human promyeloid leukemia cell line HL-60 and a
human myeloid leukemia cell line U-937 have been well
studied for the effect of vitamin D3. In HL-60 cells, vitamin
D3-induced antiproliferation and differentiation is associated
with a reduction in c-myc and pRB mRNAs (346) and an
induction of the c-fms gene, which encodes the monocyte CSF
receptor that favors differentiation (347). The principal block
to cell cycle progression in 1a,25(OH)2D3-treated human
cells occurs in G1. In HL-60 cells, the addition of
1a,25(OH)2D3 induces late G1 arrest (348) associated with
elevated levels of p27Kip1 protein, cyclin D1 (349), and p21Cip1
(350, 351) and a reduction in pRB phosphorylation. p21Cip1
was transcriptionally induced by 1a,25(OH)2D3 in a vitamin
D receptor-dependent, but not p53-dependent manner. A
vitamin D response element in the p21Cip1 promoter was also
identified. The induction of p21Cip1 was an early event, however, and not a uniform finding by all investigators, whereas
the induction of p27Kip1 protein occurred later, correlated
with the alteration in cellular phenotype (349, 352), and occurred at a posttranslational level as mRNA abundance was
unchanged (353). The main target of the elevated p27Kip1 in
this system is Cdk6 (352). The activity of Cdk2 is also downregulated, and this is associated with altered and reduced
levels of cyclin E in the kinase complex. The kinase activity
of Cdk4 was elevated, in spite of an almost complete G1
block. These data show that the functions of Cdk4 and Cdk6
are not redundant and that Cdk6 and Cdk2 activities are
regulated by 1a,25(OH)2D3 (352).
Transient overexpression of p21Cip1 and/or the related
CKI p27Kip1 in U937 cells in the absence of 1a,25(OH)2D3
results in the cell surface expression of monocyte/macrophage-specific markers, suggesting that these two CKIs were
sufficient for at least a partial induction of the differentiated
phenotype (354). Overexpression of cyclin D1 induced differentiation in the DAMI megakaryocytic cell line (355) and
in the HC11 breast cancer cell line (201), suggesting that the
abundance of the cyclin D1/p27Kip1 complex may play a
more complex role in differentiation.
6. Androgens and orphan nuclear receptors. In immature males,
androgens stimulate prostate cellular growth (356). Castration is associated with a reduction in cellular proliferation
and apoptosis in androgen-dependent epithelial cells (357).
Androgen replacement to castrated animals induces prostate
epithelial cell proliferation (356) concordant with the induction of a variety of immediate early and other genes (358). Rat
ventral prostate undergoes proliferation upon androgen replacement to castrated rats, in association with an induction
of G1 cyclins (cyclin D1, cyclin E, cyclin A), and a reduction
of these cyclins upon androgen withdrawal-induced apoptosis, although the apoptosis was p53 independent (359).
517
Cyclin D1 levels increased 9-fold, peaking at 48 h after androgen treatment (360). p27Kip1 levels increased after androgen treatment with an initial peak after 24 h of treatment.
p21Cip1 levels increased after 72 h and continued to increase
in association with a reduction in the proliferative changes
in the prostatic epithelial cells (360). In the LNCaP-FGC prostate cancer cell line, androgens increased Cdk2 and Cdk4
expression and down-regulated p16Ink4a levels (361). In the
PC3 prostate cancer cell line, which lacks AR, p16Ink4a levels
were very low, raising the possibility that the reduction in
p16Ink4a may contribute to the enhanced proliferation in prostate cells.
The growing family of orphan nuclear receptors is being
delegated roles more frequently in the regulation of cellular
differentiation and proliferation. The peroxisome proliferator-activated receptor (PPARg) binds two distinct ligands,
the synthetic antidiabetic thiazolidinediones (362, 363) and
the 15-deoxyD12,14 PGJ2 (362, 364). The induction of PPARg
during adipocyte differentiation occurs relatively early (365,
366), and retroviral overexpression of PPARg in the presence
of Pioglitazone (Pharmacia & Upjohn, Inc., Kalamazoo, MI),
a synthetic thiazolidinedione, is sufficient to induce growth
arrest and adipocyte differentiation (367). These changes
were unassociated with changes in p21Cip1 or p27Kip1 expression but were associated with a reduction in binding of
the E2F/DP proteins to an AdE2 E2F site, and it was proposed that this reduction in binding may play a role in
withdrawal of cells from the cell cycle (367).
D. Intracellular second messengers
1. cAMP. cAMP has long been known to either inhibit (368)
or induce cellular proliferation depending upon the cell type
(369). Cell cycle progression in Xenopus egg extracts is accompanied by fluctuations in cAMP levels and the activity of
PKA (370). The antiproliferative effect of cAMP has been
correlated with the induction of a protein phosphatase activity (371). The CREB and related cAMP-responsive element
modulator (CREM) proteins have specific roles in regulating
cell cycle changes. In the FRTL5 thyroid cell line, cAMP
induces cellular proliferation, and a dominant negative of
CREB caused G1 phase arrest (255). In pituitary cells, cAMP
serves as a mitogenic signal for somatotrophs. Pituitary expression of a dominant negative mutant of CREB resulted in
pituitary hypoplasia specific for the somatotrophs, in conjunction with a dwarf phenotype (372). Cyclin D1 expression
is inhibited by cAMP (177). Treatment of macrophages with
cAMP analogs induces G1 phase arrest (373), and cAMP
blocked the mitogenic effect of CSF-1, arresting the cells in
mid-G1 (100). cAMP inhibited cyclin D1 and Cdk4 expression
in CSF-1-treated macrophages in some studies (374, 375) but
not others (100). Kato et al. found that cAMP treatment of
macrophages was associated with the induction of a cyclin
D1/Cdk4 complex that was poorly phosphorylated by the
CAK. In the cAMP-treated cells, p27Kip1 levels were elevated
and immunodepletion of p27Kip1 restored the ability of CAK
to activate the cyclin D1/Cdk4 complexes (376).
Recent studies have analyzed the importance of the
changes in D-type cyclin expression observed in cAMPtreated cells. The reduction in cyclin D1 induced by cAMP
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
518
PESTELL ET AL.
appears to be critical for the inhibition of cellular proliferation in fibroblasts (377). TSH induced nuclear cyclin D3 expression in dog thyrocytes (258), and immunoneutralizing
antibodies to cyclin D3 reduced G1-S phase progression induced by TSH. Microinjection of p16 blocked TSH-induced
BrdU incorporation in FRTL5 cells, consistent with a role for
cyclin D/Cdk activity in TSH-induced mitogenesis (18). In
ovarian granulosa cells, cyclin D2 is induced by forskolin and
by FSH through a cAMP-dependent pathway (261). Although p27Kip1 levels change in response to cAMP (376), as
yet the requirement for p27Kip1 in the cytostatic effect remains to be formally established using either cells derived
from animals homozygously deleted of the p27Kip1 gene or
immunoneutralizing antibodies in cultured mammalian
cells.
The cyclin A gene is also inhibited by cAMP in a cell
cycle-dependent manner, and components of the molecular
mechanisms governing the expression of cyclin A have been
examined in several cell types. In human diploid fibroblasts
(Hs 27), cyclin A gene expression at G1/S is stimulated by 8bromo-cAMP and suppressed by the protein kinase A inhibitor H89, which delayed S phase entry in Hs 27 primary
embryo fibroblasts (378). The cyclin A gene CRE conveyed
cAMP-induced promoter activation (378). The CRE bound
CREB/CREM proteins in primary fibroblasts (378) and
ATF-1 in growing bovine aortic endothelial cells (379). In
CHO cells, inhibition of cellular proliferation by cAMP was
associated with inhibition of cyclin A promoter activity (380).
In AtT20 cells corticotroph cells, cAMP induces a cell cycle
arrest at the G2/M phase associated with an inhibition of
cyclin A expression (381). The CREM gene isoform ICER
(inducible cAMP early repressor), when overexpressed in
AtT20 cells, was capable of inhibiting G2/M phase progression and inhibited cyclin A expression. Thus, cyclin A expression is repressed in several different cell types in which
cAMP is cytostatic, and the effects appear to be mediated
through a consensus CRE site.
cAMP may also indirectly affect the cell cycle-regulatory
apparatus through an alternate mechanism affecting cyclin
degradation. cAMP also directly affects components of the
ubiquitin pathway. Anaphase-promoting complex has ubiquitin ligase activity and is required for mitotic cyclin destruction and sister chromatid separation. The 20S cyclosome
complex formation and proteolytic activity was inhibited by
the cAMP/PKA pathway (382). Together these studies suggest that both the transcriptional repression and altered protein degradation may mediate the effects of cAMP on the cell
cycle.
2. 12-O-tetradecanoylphorbol 13-acetate (TPA). The phorbol ester, TPA, activates PKC and thereby induces the activities of
many different transcription factors including AP-1, NF-kB,
p67TCF, and related Ets protein family members (383). Although induction of MAPKs and immediate early gene expression is a common finding in many different cell types, the
effect of TPA on cell cycle progression varies considerably.
Thus, TPA inhibits NIH 3T3 cell proliferation and arrests
growth of many leukemia cell lines such as the HL60 and
K562 cells, but stimulates the proliferation of Swiss 3T3 cells
(384). TPA has been shown to stimulate or inhibit different
Vol. 20, No. 4
subtypes of NIH3T3 cells (385). In both NIH3T3 cell sublines,
TPA induced immediate early gene expression. In the NIH
3T3 subline that proliferated in response to TPA (P-3T3 cells),
cyclin D1 levels were also induced. In the NIH3T3 subline
inhibited by TPA, cyclin E kinase activity was reduced.
p21Cip1 and p27Kip1 levels were not differentially affected in
the two sublines, suggesting the differential induction of
cyclin D1 may be an important distinguishing feature of the
proliferative phenotype.
In K562 cells, p21Cip1 levels were induced by TPA in a
p53-independent manner, associated with cell cycle arrest
and the induction of cellular differentiation (351). TPA induction of p21Cip1 in K5632 cells was associated with growth
arrest, and induction occurred through an AP-2 binding site
(386). AP-2 has long been known as a TPA-inducible transcription factor (387); however, the ability of AP-2 to induce
cell cycle arrest (386) raises important questions about its role
in cell cycle regulation.
IV. Cyclins and CKIs in Endocrine Tumors
Many checkpoints become deregulated during oncogenesis. In fact, restriction point-control abnormalities are an
almost universal finding in human endocrine tumors. These
abnormalities include overexpression of the cyclins (D and E)
and/or inactivation of pRB or relative reduction in the activity of the CKI by mutations or deletions. Studies in which
specific cyclins or CKI were deleted have provided insight
into the normal role for these proteins in mouse growth and
development. It is important to recognize, however, that a
molecule’s role (or lack thereof) in development does not
necessarily foreshadow or correspond to its role in tumorigenesis. Transgenic mice in which the Cdk-interactive domain of p27Kip1 was homozygously deleted developed pituitary cell tumors and gonadal growth abnormalities (388).
Homozygous deletion of the p57Kip2 gene resulted in mice
with adrenal cortical hyperplasia and cytomegaly (108).
p16Ink4a abundance is reduced in a significant proportion of
tumors (389). The incidence of abnormalities of the cyclin
D1/Cdk4/p16/pRB axis in human cancer appears to be second only to that of p53 (1). Several lines of evidence suggest
that a reduction in the abundance of the Ink4 proteins may
promote cellular proliferation and tumorigenesis. Overexpression of p16 inhibited Ras-induced foci formation in cultured cells. Homozygous deletion of the p16 gene locus,
which includes two different gene products, p16Ink4a and
p19ARF, resulted in mice with a propensity to develop lymphomas and sarcomas and enhanced tendency to develop
tumors in the presence of carcinogens (66). A similar propensity toward tumor formation was observed in mice homozygously deleted of the p19ARF gene alone, raising the
possibility that, although infrequently implicated as pathogenic in human tumorigenesis, the p19ARF gene product may
provide tumor suppressor function in the mouse (67). These
studies provided tantalizing implications for the pathogenesis of regulatory control of these endocrine organs and
fueled further investigation of the role of these CKI in human
endocrine tumors.
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
A. Parathyroid adenomas
As noted above, the human cyclin D1 gene was cloned
during molecular investigation of parathyroid adenomas in
which the PTH gene was found in a pericentromeric inversion in chromosome 11 (22) (Fig. 3). Such clonally selected
abnormalities involving the cyclin D1/PRAD1 gene provide
overwhelming evidence for PRAD19s oncogenicity in this
tissue. This is confirmed by transgenic mice overexpressing
cyclin D1 in parathyroid tissue, in which parathyroid growth
and functional abnormalities are seen (Y. Hosokawa, K. Yoshimoto, and A. Arnold, unpublished). In parathyroid neoplasms, overexpression of the wild-type PRAD1 sequence,
rather than mutational activation, appears to be the predominant mechanism by which PRAD1 exerts its oncogenic action (390). Overexpression of cyclin D1 was found in 18% of
parathyroid neoplasms in one study (391). Cyclin D1 overexpression is a feature of typical parathyroid adenomas (391).
Hyperplasias had 3-fold more p27Kip1-positive cells than
parathyroid adenomas, suggesting that p27Kip1 immunostaining may be useful in distinguishing between these two
conditions (392). Deletions of the p16Ink4a and p15Ink4d genes
occur uncommonly, if ever, in parathyroid adenomas (393).
In one study the large majority of parathyroid carcinomas
showed allelic losses on 13q that included the RB gene, associated with significantly reduced or absent nuclear staining for pRB, whereas none of the benign parathyroid adenomas examined contained abnormal pRB staining (394).
Frequent allelic loss on 13q has been confirmed in aggressive
parathyroid tumors (395), but it remains to be determined
whether pRB itself is a functionally relevant target of such
deletions.
B. Thyroid cancer
Oncogenes known to disrupt the cyclin D1/pRB axis have
been shown to induce thyroid tumors. When targeted to the
thyroid gland by the bovine thyroglobulin promoter, the
SV40 large T antigen induced thyroid adenocarcinoma (396),
and the E7 protein induced papillary and follicular thyroid
cancer (397). p53 Is frequently mutated in anaplastic thyroid
carcinomas, and p21Cip1 is a downstream effector of p53. It
is conceivable that genetic defects of genes downstream in
the p53 pathway could also be oncogenic. In analysis of
thyroid tissues including adenomas and carcinomas by one
group, normal thyroid follicles and adenomas rarely were
immunopositive for p21Cip1; however 30 of 93 carcinomas
studied were positive for p21Cip1. There was no correlation
between p21Cip1 immunopositivity and p53 status. The incidence of p21Cip1 status was unrelated to thyroid carcinoma
subtype when undifferentiated, poorly differentiated, and
well differentiated tumors were compared. There was also no
correlation between p21Cip1 status and clinical parameters
(398). In other studies p21Cip1 abnormalities were not found
in thyroid adenomas; however, 12% of the papillary carcinomas harbored p21Cip1 mRNA deletions of exon 2, without
evidence for gene deletion (399) and therefore of uncertain
pathogenetic importance. A polymorphism in the p15Ink4b
gene was noted to occur more frequently in papillary and
medullary thyroid cancer compared with normal subjects,
519
although the functional significance of this polymorphism in
the tumor suppressor function remains undetermined (400).
The role of other cell cycle-regulatory proteins such as cyclin
D1 and the other CKI as independent prognostic indicators
in thyroid cancer, as previously performed in breast and
prostate cancer, remains to be assessed.
C. Pituitary tumors
Homozygous deletion of the p27Kip1 gene resulted in
larger animals with organomegaly and intermediate lobe
pituitary tumors, suggesting the p27Kip1 product played a
role in inhibiting normal cellular growth, particularly in the
pituitary (104, 105). Among 31 sporadic and 2 familial pituitary adenomas, PCR-single-strand conformation polymorphism analysis detected three polymorphic changes but no
tumor-specific mutations of the p27Kip1 gene (401). p27Kip1
levels were reduced in neoplastic pituitary tissue (392), and
Jin et al. (402) found that p27Kip1 levels were normally high
in pituitary cells but protein levels were decreased in adenomas and carcinomas (402).
Transgenic mice heterozygous for a pRB mutation developed pituitary corticotrope intermediate lobe tumors (403,
404). These tumors were in part dependent upon inhibition
of E2F-1 function as mating of mice nullizygous for E2F-1
with the pRB mutant mice resulted in offspring with a reduced prevlance of pituitary tumors (405). In studies of human pituitary tumors, however, pRB mutations were not
found (406, 407), and in aggressive tumors the loss of heterozygosity was found at sites telomeric and centromeric to
the pRB locus (408). Functional inactivation of pRB through
overexpression of G1 cyclins in pituitary tumors remains to
be formally examined. As abnormalities of either the pRB or
p53 axis are observed almost uniformly in human tumors of
all types (1), it was surprising that p53 mutations were not
found in pituitary tumors (409). In transgenic mice homozygously deleted of the p18INK4c gene, hyperplasia of the intermediate and anterior pituitary gland was found as early
as 4 weeks (410). Intermediate-lobe pituitary tumors staining
for ACTH subsequently developed with nearly complete
penetrance
(410). Mating of the p18Ink4c2/2 and the
Kip12/2
mice resulted in an acceleration of the pituitary
p27
pathology consistent with the model that these two CKI work
in distinct parallel pathways. Thus, to summarize, pituitary
tumors in the mouse result from deletion of genes for specific
CKI that are expressed well in the normal murine pituitary
(p18Ink4c and p27Kip1), but are not found with deletion of
other CKI (p16Ink4a, p21Cip1, p57Kip2). In all, the role of cyclin/
CKIs in human pituitary tumors as prognostic markers or
mechanistic abnormalities remains relatively poorly understood (411).
D. Adrenal tumors
As noted above, homozygous deletion of the p57Kip2 gene
resulted in mice that developed adrenal cortical hyperplasia
and cytomegaly (109). Although the low frequency of p57Kip2
mutations in patients with Beckwith-Wiedemann syndrome
in conjunction with the frequent disruption of the K(v)LQT1
gene in patients with chromosomal rearrangements sug-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
520
PESTELL ET AL.
gested that Beckwith-Wiedemann syndrome involves disruption of multiple independent 11p15.5 genes (412), these
results stimulated interest in the role of p57Kip2 in adrenal
tumorigenesis. Expression of p57Kip2 was restricted in the
mouse to the fetal adrenal. In normal human adrenals, however, p57Kip2 mRNA (and H19 RNA) was abundantly expressed and was found in adrenocortical adenomas from
patients with Cushing’s or Conn’s syndrome. In most adrenocortical carcinomas and virilizing adrenal adenomas, very
low levels of both p57Kip2 and H19 RNAs were observed
(413). These findings remain consistent with a model in
which p57Kip2 may play a role in the inhibition of abnormal
adrenal cellular proliferation.
E. Ovarian and testicular cancer
Assessment of components of the CKI/E2F axis in ovarian
and testicular tumors revealed surprising results. Based on
epithelial cell tumor analysis, it was anticipated that CKI
levels would be reduced and E2F-1 levels may be increased.
Surprisingly, p16Ink4a mRNA and protein levels were significantly elevated in 28/32 ovarian tumors (414). When
p16Ink4a gene deletions and rearrangements were sought in
21 ovarian and 42 testicular tumors, no abnormalities other
than polymorphisms were found (415). These findings were
somewhat counterintuitive, as overexpression of E2F-1 typically induces S-phase progression and apoptosis. p21Cip1
levels were reduced, however, in ovarian cancer (416). The
homozygous deletion of the p27Kip1 gene resulted in animals
with testicular and ovarian cell hyperplasia (104, 105), raising
the possibility that p27Kip1 may normally inhibit testicular
and ovarian cell proliferation. The testes of mice homozygously deleted of the E2F-1 gene underwent atrophy (417,
418), although the mechanism is currently not defined. These
findings suggest that the role of p21Cip1 and p27Kip1 as ovarian and testicular cell tumor suppressors warrants further
investigation.
Homozygous deletion of cyclin D2 was associated with
selective abnormalities of gonadal development (261). The
association with tumor formation is much less frequent for
cyclin D2 and cyclin D3 than with cyclin D1, and cyclins D2
and D3 have not yet proven to be human oncogenes. Cyclin
D2 overexpression has been observed in a subset of gonadal
tumors in which cyclin D1 levels were unaltered (261). Cyclin
D2 levels, which were normally undetectable, were increased
in male germ cell tumors (419). The levels of cyclin D2 were
inversely correlated with the level of tumor differentiation.
p21Cip1 levels were inversely correlated with cyclin D2 levels
(419). Cyclin D2 was associated with Cdk6 and Cdk4 in the
tumors (419). At present, among the D type cyclins, cyclin D2
overexpression appears to be linked specifically to a subset
of gonadal tumors.
F. Mechanisms of oncogenic transformation by cyclin D1
The cyclin D1 gene maps to the region of chromosome
band 11q13, which has been frequently found in tumorassociated translocations. Evidence from several laboratories
has indicated that the PRAD1 is the BCL-1 (B-cell lymphoma/leukemia 1) protooncogene (420 – 422) critical in the
Vol. 20, No. 4
pathogenesis of mantle cell lymphoma. The cyclin D1 part of
the 11q13 chromosomal band was also found to be amplified
in numerous other types of malignancy including breast,
squamous cell, esophageal cancers, and a smaller number of
bladder, hepatocellular, and non-small cell lung cancers (9,
423). The association with tumor formation is much less
frequent for cyclin D2 and cyclin D3 than with cyclin D1.
Cyclin D2 overexpression has been observed in a subset of
gonadal tumors (261), and cyclins D2 and D3 have also been
implicated in chronic lymphocytic leukemia and malignant
lymphoma. The relative importance of structural differences
in the D-type cyclin promoters to their tumorigenic or developmental roles is an area of active investigation.
Cyclin D1 mRNA is overexpressed in many different neoplasms including breast, esophageal, hepatic, squamous cell
carcinoma of the head and neck, and melanoma. The prevalence of cyclin D1 overexpression is particularly high in
mantle cell lymphomas and breast cancers. Some 30 – 45% of
human breast cancers and 70 –100% of breast tumor cell lines
have increased cyclin D1 expression. Although in head and
neck cancers, the increase in cyclin D1 abundance is usually
associated with cyclin D1 gene amplification, gene amplification can explain only some of the overexpression found in
breast cancers, suggesting that additional factors contribute
to the increased abundance of cyclin D1 (1). Immunohistochemical analysis of cyclin D1 abundance provides independent prognostic information in squamous cell carcinoma of
the head and neck (424) and pancreatic cancer (425), in which
the overexpression of cyclin D1 is associated with poor prognosis. Some (426) but not all studies (427) suggest cyclin D1
abundance may provide useful prognostic information in
breast cancer. Immunohistochemical analysis of cyclin D1
abundance may be useful in predicting relapse in patients
with good-prognosis breast cancer (428). The recent development of cyclin D1 enzyme-linked immunosorbent assays
of tumors may provide information with higher predictive
value for patient management in breast and other cancers but
remains to be fully evaluated.
Cyclin D1 alone is not capable of transforming primary
cultured cells but is capable of cooperating in classical assays
of transformation with other known oncogenes. Primary
baby rat kidney cells (BRK) cells, like rat embryo fibroblasts,
cannot be transformed by oncogenes such as E1A or Myc
without a collaborating oncogene such as Ras. Although
cyclin D1 alone did not transform BRK cells, it enhanced
3-fold the transformation efficiency of a mutant E1A that was
incapable of binding pRB (429). The complementation of
cyclin D1- transforming ability required binding to its catalytic partner, Cdk4, but did not require the pRB-binding
domain (429).
The oncogenic capacity of cyclin D1 has been investigated
in transgenic mice. Overexpression of cyclin D1 in the mammary glands of transgenic mice resulted in mammary gland
tumor formation, (430) supporting cyclin D1’s role as a
“driver oncogene” on the 11q13 amplicon in breast cancer.
Coexpression of cyclin D1 and N-Myc or L-Myc under control of an Em-directed enhancer sequence in double transgenic mice revealed a strong cooperative effect in the rapid
development of clonal pre-B and B cell lymphomas (431, 432).
Transgenic mice in which the cyclin D1 cDNA was linked to
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
the Epstein-Barr virus promoter developed squamous epithelial cell dysplasia in the tongue, esophagus, and forestomach, sites demonstrated to express the transgene (433).
Similar types of transgenic experiments demonstrated that
keratin promoter-directed overexpression of cyclin D1 in
epithelial cells could induce epithelial cell hyperplasia (434).
Because of the clear evidence for the oncogenicity of cyclin
D1 in vivo, recent studies have further examined the requirement for cyclin D1 in cellular transformation. In cultured
fibroblasts, antisense cyclin D1 reduced Ras-induced transformation (435). When transgenic mice with skin tumor induced by Ras were mated with mice homozygously deleted
of the cyclin D1 gene, the prevalence of skin tumors was
significantly decreased, providing further supporting evidence for a critical role of cyclin D1 in Ras-induced transformation (436). Stable integration of an antisense cyclin D1
cDNA in a transplantable colonic cancer cell line inhibited
tumor formation of the implanted cells in nude mice (437).
These results provide encouragement to investigators seeking important targets for alternative molecular or pharmacological tumor therapy, as the cyclin D1/p16/pRB axis is
frequently perturbed in tumors.
As noted above, a variety of oncogenes induce overexpression of cyclin D1. Several different protooncogenes induce cyclin D1 abundance and cyclin D1 promoter activity
including activating mutants of Ras, pp60src, Rac, Neu, and
Dbl and overexpression of SV40 small t antigen (187, 438 –
440). In some tumors, the mRNA cap-binding protein (eukaryotic initiation factor 4E [eIF- 4E]) increases cyclin D1
protein levels likely at a posttranscriptional level (441, 442).
The mechanism by which cyclin D1 overexpression induces
cellular transformation is distinct from many other oncogenes. The cyclin D1 protein and coding sequence from tumors examined to date are normal, suggesting it is the overexpression of cyclin D1 per se that is responsible for the
formation of tumors (7, 9). The weight of evidence supports
the proposal that overexpression of wild-type cyclin D1 contributes to transformation, in large part, through phosphorylation and inactivation of the pRB protein (5), although this
has not been well examined in complex in vivo systems.
Additional mechanisms may well contribute to the transforming ability of cyclin D1 including the sequestration of
CKI, such as p27Kip1, and perhaps the ability to bind other
proteins through Cdk-independent mechanisms.
The coding sequence derived from some immortal cell
lines had substitutions that would lead to amino acid
changes, but the possibility that these amino acid changes
may lead to activating mutations has not been directly assessed. An alternatively spliced form of cyclin D1 that lacks
the carboxy terminus involved in targeting cyclin D1 for
destruction has been described (28). The relatively long 39noncoding region of cyclin D1 is also occasionally truncated
in tumor cell lines. Overexpression of this alternate splice
form appears to differentially inhibit G0-G1 phase progression compared with the wild type in U87 mG cells (443).
Cyclin D1 overexpression is frequently associated with gene
amplification at other distinct loci, and it has been suggested
that cyclin D1 overexpression may contribute to gene amplification and genomic instability that occurs during cellular
transformation (444).
521
Indirect evidence for other mechanisms contributing to
cellular transformation by cyclin D1, however, do exist. Cyclin D1 interacts directly with other proteins to affect gene
transcription, which in turn may contribute to the transforming phenotype. For example, cyclin D1 binds the ER, enhancing estrogen-dependent enhancer activity (114, 115). As
noted above, this effect is independent of the Cdk4 function
of cyclin D1. Cyclin D1 also affects activity of V-Myb (117)
and binds a Myb-related protein (116). Although a formal
assessment has not yet been made, it is tempting to speculate
that these protein-protein interactions may also contribute to
cyclin D1’s tumorigenic properties.
G. CKIs in treatment of endocrine disease
Because major changes in the activity of the Cdks are
found in the majority of tumors, enzymatic screening has
been used to uncover chemical inhibitors for tumor therapy
(445). The first generation of these inhibitors have been studied in some detail and are being used in clinical trials of
tumor therapy. Although there are several different theoretical approaches to identification of CKIs, the majority of these
drugs were identified as inhibitors of Cdk catalytic activity.
Molecular interaction-based screening is beginning to identify inhibitory peptides (446), and the resolution by crystal
structure of the interfaces between the CKI and their target
proteins provides a logical basis for ongoing studies. Although the ability of these compounds to preferentially inhibit Cdk activity compared with other kinases such as PKC
has been suggested, further studies need to be performed
using synthetic substrates including pRB in cultured cells.
The seven types of CKIs currently described in detail include
1) staurosporine and UCN-01 (two microbial alkaloids); 2)
butyrolactone; 3) flavopiridol and L868276 (derived from an
Indian plant extract); 4) 9-hydroxy-ellipticine (derived from
the plant Ochrosia ellipticia); 5) suramin (a naturally occurring
glycosaminoglycan); 6) olomoucine, roscovitine, and isopentenyladenine; and 7) peptides, some of which are derived
from the sequence of the naturally occurring CKI described
above (445). Flavoperidol was the first specific CKI to undergo clinical trials on a spectrum of malignancies. Other
than the peptides, these inhibitors were all derived from
natural sources. It is hoped that the preferential activity of
some of these inhibitors for particular Cdks may allow selective use for tumor therapy.
V. Transcription Factors Regulating the Cell Cycle
Several different laboratories have provided important insights into the transcription factors that regulate the G1 cyclins and the CKI, and the role of these DNA sequences in
hormone signaling has been investigated. The promoter
structures of the cyclin D1, D2, D3, and cyclin A promoters
are shown schematically in Fig. 8B with the transcription
factor-binding sites designated. Transcription factors binding to the cyclin D1 (26), cyclin A (447), and cyclin E promoters (448) were identified through a combination of transient expression studies, mutational analyses, and
electrophoretic mobility shift assays. Specific components of
the basal transcription apparatus are required for normal cell
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
522
PESTELL ET AL.
cycle progression and, in some cell types, have been shown
to link directly to the induction of cyclin gene expression.
TAFII250 is a core component of transcription factor IID
(TFIID) and binds to a number of other subunits including
TATA-binding protein (TBP) and other transcription-activating factors (TAFs). Mutant strains of Saccharomyces cerevisiae, containing temperature-sensitive mutants of
yTAFII145, the yeast homolog of human TAFII250, have been
shown to arrest at the restrictive temperature (449). The
mutant hamster cell line, tsBN462, exhibits cell cycle arrest at
the restrictive temperature due to a point mutation in the
TAFII250/CCG1 gene (450). This cell line fails to progress out
of G0 into G1 and was found to express reduced levels of
cyclin D1, cyclin A, and cyclin E and increased levels of
p21Cip1 and p27Kip1 (450). The arrest may be due to the reduction in cyclin D1 and cyclin A as cyclin D1 was shown to
be directly induced by TAFII250 (451). The cyclin A promoter
was also a direct transcriptional target, with induction by
TAFII250 occurring through ATF- binding sites consisting of
ATF1, CREB1, and ATFa2 in HeLa cell extracts (451).
Cyclin D1 mRNA levels are induced primarily at the transcriptional level. Recent studies have demonstrated a role for
the Ras/MAPK intracellular signaling pathway in growth
factor induction of cyclin D1 expression and promoter activity and have identified transcriptional targets at the distal
end of these pathways (Fig. 8). Overexpression of either
human or Xenopus ERK cDNAs (187, 250) and specific activating mutations of the ERK kinase (439, 452) induced cyclin
D1 promoter activity. The induction of ERK activity likely
plays an important role in the induction of cyclin D1 by
oncogenic mutations of p21Ras (187), Rac (438), members of
the dbl family (440), and Neu (453). The relative abundance
of cyclin D1 is dependent upon the relative abundance of
both the MAPKs and their MAPK phosphatases such as
mitogen-activated MKP-1 and PP2A, both of which reduce
MAPK activity. MKP-1 is known to have cell cycle-inhibitory
activity, and overexpression of MKP-1 inhibited cyclin D1
(439). Inhibition of the MAPK phosphatase PP2A by the SV40
small t antigen induced cyclin D1 levels and CD1K activity
(439). The domain of SV40 small t required for PP2A binding
was required for induction of cyclin D1. These results suggest
that relative balance of the MAPKs and their phosphatases
determine the abundance of cyclin D1.
Analysis of transcriptional regulation of the cyclin D1 gene
has led to the identification of distinct binding sites at the
distal end of oncogenic signaling pathways. This structure of
the cyclin D1 regulatory region may allow for collaborative
interactions between oncogenic stimuli. c-Jun activation of
the cyclin D1 promoter (187, 454) required an activator protein 1 (AP-1) (c-Fos/c-Jun) binding site at 2953 (187). This
AP-1 binding site is the same region shown to be involved
in AII-induced mitogenesis (250) (Fig. 8B). Sequences at 258
bind CREB in trophoblastic cells (439), ATF-2 in breast cancer
cells (455) and chondrocytes (456), and Fos in primary fibroblasts (457). The cyclin D1 gene was activated by b-catenin (458, 459) through DNA sequences shown to bind a
lymphoid enhancer factor-1 (LEF-1)/b-catenin heterodimer
(458). Constitutively active STAT5 induced the cyclin D1
promoter through a STAT-binding site at 2481 (460). pRB
was shown to induce cyclin D1 expression, and Sp-1 and E2F
Vol. 20, No. 4
FIG. 8. Transcriptional regulation of the G1 cyclins. A, Several different growth factors and oncogenes have been shown to induce cyclin
D1 expression through the cyclin D1 promoter. Induction through Ras
and Rac requires ERK-independent signaling pathways (438). The
ERK pathway is also capable of directly activating cyclin D1 abundance (438). B, Schematic representation of the G1 cyclin promoters.
Several transcription factors binding to the promoter have been identified through mutagenesis and electrophoretic mobility shift assays.
The cyclin D1 promoter binds AP-1 proteins at 2953 (250), E2F/p107
proteins at 2145 (461), Sp1/Sp-3 at 2130 (461), and CREB/CREM
proteins at 254 (439), and Ets proteins function to modulate proximal
promoter activity (187). DNA sequences have been identified in the
cyclin D2 and D3 promoter, but nuclear protein binding to these sites
have yet to be shown (468). The human cyclin A promoter 2237 to
2214 binds ATF1, CREB1, and ATFa2 and enhances activity in a
TAFII250-dependent manner (451), a CRE at 280 to 273 (378), and an
E2F site at 237/232 (478, 479). The murine cyclin A promoter contains
a CRE consensus at 230 and a GC-rich sequence at 114, which plays
a role in negative regulation during cell cycle progression (480).
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
proteins bind to the proximal promoter modulating regulation by pRB and the E2F transcription factors (Fig. 8B) (461).
In several of these analyses, cyclin D1 abundance was rate
limiting in the proliferative or transforming phenotype of the
transcription factor examined (456, 457). Thus, cyclin D1
levels were reduced in the chondrocytes of the ATF-22/2
chondrocytes and the c-fos/fosB2/2 mouse embryo fibroblasts in conjunction with reduced levels specifically of cyclin
D1. The abundance of cyclin D1 was also critical in interleukin-3 and STAT5-dependent cell growth (460). It has been
proposed therefore that the relative abundance of the transcription factors binding to these sites in the cyclin D1 promoter may contribute to the ability of a particular cell type
to respond to a given mitogenic signal.
Controversy surrounds the role of c-Myc in regulating
cyclin D1 expression (462, 463), and the role of c-Myc in
hormone signaling is not well understood. Cyclin D1 levels
are frequently increased in pRB-overexpressing tumors, and
overexpression of pRB induced cyclin D1 levels (464). c-Myc
induces cyclin D1-kinase activity (462) or protein levels in
some (465) but not other studies (463). Although in most
circumstances, cyclin D1 abundance is regulated at the level
of enhanced transcription, a contribution to cyclin D1 abundance at posttranscriptional level may mediate the effect of
c-Myc on cyclin D1. The relative abundance of pRB and p53
in the cell may determine the effect of a particular signaling
pathway on cyclin D1 abundance. Induction of p53 was
associated with increased cyclin D1 abundance, and the induction occurred independently of the p21Cip1 status of the
cell in one study (466). However other investigators have
shown that p53 induction of cyclin D1 is through p21Cip1
(466). The requirement for cyclin D1 in p53-mediated arrest
may reflect the ability of cyclin D1 to induce cell cycle arrest
in specific cell types including normal human diploid fibroblasts (11, 30, 467). p300, A coactivator protein that regulates
a diverse array of transcription factors, regulates cyclin D1
gene expression and associates with it in multiprotein complexes (Ref. 140 and C. Albanese and R. G. Pestell, submitted). The interaction between cyclin D1 and p300 may therefore be of considerable importance, allowing cyclin D1 to
regulate a broad array of genes during cell cycle progression.
Ongoing studies are attempting to unravel the contribution
of these additional protein-protein interactions to the diverse
role of cyclin D1 in cellular differentiation, proliferation, and
transformation.
The human cyclin D2 and cyclin D3 promoter regions have
been sequenced (468). The cyclin D2 promoter contains DNA
sequences homologous to binding sites for AP-2, PUF, and
Sp1 binding (468). The human cyclin D3 contains sequences
resembling AP-2-, Sp-1-, and CTF-binding sites within the
proximal 2366 to 2167-bp region involved in basal enhancer
activity. The rat (469) and mouse (470) cyclin D3 promoters
also contain Sp-1- and AP-2-like sequences in the proximal
2350 bp, although the functional significance and the nature
of the proteins binding to these sequences remain to be
determined.
The human cyclin A gene contains a CRE that contributed
to the timing of cyclin A expression in fibroblasts and was
shown to bind CREB/CREM proteins in one study (378) and
ATF-1 in another (380). The TGFb1-induced down-regula-
523
tion of cyclin A promoter activity appeared to be mediated
via the activating transcription factor (ATF) site (379). This
effect occurred in association with reduced CREB phosphorylation bound to the proximal ATF/CRE-binding site (471).
Because of the distinct temporal profile of induction of the
cyclins at the transcriptional level and the distinct responses
of these genes to different trophic stimuli, it is likely that
focused chimeric promoter swaps will provide important
information about the mechanisms governing these differential responses.
VI. Conclusion
Dramatic advances have occurred in the last 10 years in
our understanding of cell cycle abnormalities in cancer. Attention has more recently turned to the role of cell cyclecontrol proteins in the normal nondividing cell in vivo. Investigations over the last few years have culminated in a new
understanding of the fundamental mechanisms by which
hormone signals modulate cellular phenotype through interactions with the cell cycle machinery. The ongoing challenge remains the identification of unifying themes within
these apparently complex schemes. Hormones may now be
thought of as regulating cell cycle-control proteins through
distinct mechanistic paradigms (Figs. 4 – 6). The focus of
these recent studies has been the role of altered phosphorylation in regulating transcriptional activity and gene expression. Attention is now turning to the role of acetylation
in the function of transcription factors and cell cycle-regulatory proteins (227, 472, 473). An understanding of the role
of acetylation in hormone signal transduction will likely
become increasingly important. Historically the management of endocrine disease has been directed at modification
of the hormone secreted. Ongoing studies of hormonal regulation of cell cycle proteins may lead to the identification of
common downstream targets responsible for regulating hormonal effects. It will be of enormous value to endocrinologists if targeted drug design can be used to treat complications of endocrine disease through usurping these final
common pathways. In addition, the systematic analysis and
identification of cell cycle abnormalities in endocrine tumors
may provide a rational basis for alternate forms of tumor
therapy.
Acknowledgments
We thank Drs. R. DePinho, M. Pagano, and E. R. Stanley for their
reading and helpful suggestions on this article.
Note Added in Proof
During the publication of this article, cyclin D1 was shown to be
transcriptionally regulated by oncogenic mutations in Stat3 (481) as well
as by NF-kB through a consensus NF-kB binding site at -58 of the cyclin
D1 promoter (482).
References
1. Sherr CJ 1996 Cancer cell cycles. Science 274:1672–1677
2. Sherr CJ, Roberts JM 1995 Inhibitors of mammalian G1 cyclindependent kinases. Genes Dev 9:1149 –1163
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
524
PESTELL ET AL.
3. Sherr CJ 1994 G1 phase progression: cycling on cue. Cell 79:551–555
4. Elledge SJ 1996 Cell cycle checkpoints: preventing an identity
crisis. Science 274:1664 –1671
5. Weinberg RA 1995 The retinoblastoma protein and cell cycle control. Cell 81:323–330
6. Arnold A 1995 The cyclin D1/PRAD1 oncogene in human neoplasia. J Invest Med 43:543–549
7. Reutens AT, Watanabe G, Shambaugh III GE, Pestell RG 1997
Cyclin D1 and the cyclin dependent kinase regulatory proteins in
tumorigenesis and differentiation. Einstein Q J Biol Med 14:3–14
8. Cordon-Cardo C 1995 Mutation of cell cycle regulators. Am J
Pathol 147:545–560
9. Motokura T, Arnold A 1993 Cyclin D and oncogenesis. Curr Opin
Genet Dev 3:5–10
10. Hunter T 1993 Oncogenes and cell proliferation. Curr Opin Genet
Dev 3:1– 4
11. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G 1993 Cyclin
D1 is a nuclear protein required for cell cycle progression in G1.
Genes Dev 7:812– 821
12. Quelle DE, Ashmun RA, Shurtleff SA, Kato J-y, Bar-Sagi D,
Roussel MF, Sherr CJ 1993 Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev 7:1559 –
1571
13. Jiang W, Kahn SM, Zhou P, Zhang Y-J, Cacace AM, Infante AS,
Doi S, Santella RM, Weinstein IB 1993 Overexpression of cyclin
D1 in rat fibroblasts causes abnormalities in growth control, cell
cycle progression and gene expression. Oncogene 8:3447–3457
14. Resnitzky D, Gossen M, Bujard H, Reed SI 1994 Acceleration of
the G1/S phase transition by expression of cyclins D1 and E with
an inducible system. Mol Cell Biol 14:1669 –1679
15. Musgrove EA, Lee CS, Buckley MF, Sutherland RL 1994 Cyclin
D1 induction in breast cancer cells shortens G1 and is sufficient for
cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci
USA 91:8022– 8026
16. Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J
1994 Cyclin D1 protein expression and function in human breast
cancer. Int J Cancer 57:351–361
17. Xiong W, Pestell RG, Watanabe G, Rosner MR, Hershenson MB
1997 Cyclin D1 is required for S-phase traversal in bovine tracheal
myocytes. Am J Physiol 272:L1205–L1210
18. Lukas J, Bartkova J, Bartek J 1996 Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin
D-cyclin dependent kinase-pRb-controlled G1 checkpoint. Mol Cell
Biol 16:6917– 6925
19. Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J 1995 Cyclin D1
is dispensible for G1 control in retinoblastoma gene-deficient cells
independently of Cdk4 activity. Mol Cell Biol 15:2600 –2611
20. Finley RL, Thomas BJ, Zipursky SL, Brent R 1996 Isolation of
Drosophila cyclin D, a protein expressed in the morphogenetic furrow before entry into S phase. Proc Natl Acad Sci USA 93:3011–3015
21. Orr-Weaver TL 1994 Developmental modification of the Drosophila
cell cycle. Trends Genet 10:321–327
22. Arnold A, Kim HG, Gaz RD, Eddy RL, Fukushima Y, Byers MG,
Shows TB, Kronenberg HM 1989 Molecular cloning and chromosomal mapping of DNA rearranged wth the parathyroid hormone
gene in a parathyroid adenoma. J Clin Invest 83:2034 –2040
23. Motokura T, Bloom T, Kim HG, Juppner H, Ruderman JV, Kronenberg HM, Arnold A 1991 A novel cyclin encoded by a bcl1linked candidate oncogene. Nature 350:512–515
24. Xiong Y, Connolly T, Futcher B, Beach D 1991 Human D-type
cyclin. Cell 65:691– 699
25. Matsushime H, Roussel MF, Ashmun RA, Sherr CJ 1991 Colonystimulating factor 1 regulates novel cyclins during the G1 phase of
the cell cycle. Cell 65:701–713
26. Motokura T, Arnold A 1993 The PRAD1/Cyclin D1 proto-oncogene: genomic organization, 59 DNA sequence, and sequence of a
tumor-specific rearrangement breakpoint. Genes Chromosomes
Cancer 7:89 –95
27. Rao SS, Chu C, Kohtz DS 1994 Ectopic expression of cyclin D1
prevents activation of gene transcription by myogenic helix-loophelix regulators. Mol Cell Biol 14:5259 –5267
28. Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD,
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
Vol. 20, No. 4
Heighway J 1995 Alternate splicing produces a novel cyclin D1
transcript. Oncogene 11:1005–1011
Diehl JA, Zindy F, Sherr CJ 1997 Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the
ubiquitin-proteasome pathway. Genes Dev 11:957–972
Pagano M, Theodorus AM, Tam SW, Draetta GF 1994 Cyclin
D1-mediated inhibition of repair and replicative DNA synthesis in
human fibroblasts. Genes Dev 8:1627–1639
Lukas J, Bartkova J, Welcker M, Petersen OW, Peters G, Strauss
M, Bartek J 1995 Cyclin D2 is a moderately oscillating nucleoprotein required for G1 phase progression in specific cell types. Oncogene 10:2125–2134
Sherr CJ 1993 Mammalian G1 cyclins. Cell 73:1059 –1065
Kiyokawa H, Busquets X, Powell CT, Ngo L, Rifkind RA, Marks
PA 1992 Cloning of a D-type cyclin from murine erythroleukemia
cells. Proc Natl Acad Sci USA 89:2444 –2447
Mumberg D, Wick M, Burger C, Haas K, Funk M, Muller R 1997
Cyclin ET, a new splice variant of human cyclin E with a unique
expression pattern during cell cycle progression and differentiation. Nucleic Acids Res 25:2098 –2105
Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM 1996
Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by Cdk2 binding and cyclin phosphorylation. Genes Dev
10:1979 –1990
Won KA, Reed SI 1996 Activation of cyclin E/Cdk2 is coupled to
site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J 15:4182– 4193
Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei
MC, Dowdy SF 1997 Hypo-phosphorylation of the retinoblastoma
protein (pRb) by cyclin D: Cdk4/6 complexes results in active pRb.
Proc Natl Acad Sci USA 94:10699 –10704
Dowdy SF, Latek RR, Nagahara H, Van Dyk LF 1996 G1 phase
hypo-phosphorylation of the retinoblastoma protein: targeting to
specific transcription factors. Proc Annu Meet Am Assoc Cancer
Res 37:A24
Lukas J, Muller H, Bartkova J, Spitkovsky D, Kjerulff AA,
Jansen-Durr Srauss M, Bartek J 1994 DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve
the cell’s requirement for cyclin D1 function in G1. J Cell Biol
125:625– 638
Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin
K, Reed SI, Bartek J 1997 Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev 11:1479 –1492
Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano
M 1995 Human cyclin E, a nuclear protein essential for the G1-to-S
phase transition. Mol Cell Biol 15:2612–2624
Resnitzky D, Reed SI 1995 Different roles for cyclins D1 and E in
regulation of the G1-to-S transition. Mol Cell Biol 15:3463–3469
Kelly B, Wolfe KG, Roberts JM 1998 Identification of a substratetargeting domain in cyclin E necessary for phosphorylation of the
retinoblastoma protein. Proc Natl Acad Sci USA 95:2535–2540
Zhao J, Dynlacht B, Imai T, Hori T, Harlow E 1998 Expression of
NPAT, a novel substrate of cyclin E-Cdk2, promotes S-phase entry.
Genes Dev 12:456 – 461
Pines J, Hunter T 1990 Human cyclin A is adenovirus E1A-associated protein p60 and behaves differently from cyclin B. Nature
346:760 –763
Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G 1992
Cyclin A is required at two points in the human cell-cycle. EMBO
J 11:961–971
Krude T, Jackman M, Pines J, Laskey RA 1997 Cyclin/Cdkdependent initiation of DNA replication in a human cell-free system. Cell 88:109 –119
Dynlacht BD, Flores O, Lees JA, Harlow E 1994 Differential regulation of E2F trans-activation by cyclin/Cdk2 complexes. Genes
Dev 8:1772–1786
Krek W, Xu G, Livingston DM 1995 Cyclin A-kinase regulation of
E2F-1 DNA binding function underlies suppression of an S phase
checkpoint. Cell 83:1149 –1158
Krek W, Ewen ME, Shirodkar S, Arany Z, Kaelin Jr WG, Livingston DM 1994 Negative regulation of the growth-promoting
transcription factor E2F-1 by a stably bound cyclin A-dependent
protein kinase. Cell 78:161–172
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
51. Bai C, Richman R, Elledge S 1994 Human cyclin F. EMBO J 13:
6087– 6098
52. Horne MC, Goolsby GL, Donaldson KL, Tran D, Neubauer M,
Wahl AF 1996 Cyclin G1 and cyclin G2 comprise a new family of
cyclins with contrasting tissue-specific and cell cycle-regulated expression. J Biol Chem 271:6050 – 6061
53. Morita N, Kiryu S, Kiyama H 1996 p53-Independent cyclin G
expression in a group of mature neurons and its enhanced expression during nerve regeneration. J Neurosci 16:5961–5966
54. Lew J, Huang Q-Q, Qi Z, Winkfein RJ, Aebersold R, Hunt T,
Wang JH 1994 A brain-specific activator of cyclin-dependent kinase
5. Nature 371:423– 426
55. Tsai LH, Delalle I, Caviness VS, Chae T, Harlow E 1994 p35 Is a
neural specific regulatory subunit of cyclin-dependent kinase 5.
Nature 371:419 – 423
56. Poon RY, Lew J, Hunter T 1997 Identification of functional domains in the neuronal Cdk5 activator. J Biol Chem 272:5703–5708
57. Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai L-H 1997 Mice
lacking p35, a neuronal specific activator of Cdk5, display cortical
lamination defects, seizures and adult lethality. Neuron 18:29 – 42
58. Xiong W, Pestell RG, Rosner MR 1997 Role of cyclins in neuronal
differentiation of immortalized hippocampal cells. Mol Cell Biol
17:6585– 6597
59. Dunphy WG 1994 The decision to enter mitosis. Trends Cell Biol
4:202–207
60. King RW, Deshaies RJ, Peters JM, Kirschner MW 1996 How
proteolysis drives the cell cycle. Science 274:1652–1659
61. King RW, Jackson PK, Kirschner MW 1994 Mitosis in transition.
Cell 79:563–571
62. Hunter T, Pines J 1994 Cyclins and cancer II: cyclin D and Cdk
inhibitors come of age. Cell 79:573–582
63. Quelle DE, Zindy F, Ashmun RA, Sherr CJ 1995 Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83:993–
1000
64. Quelle DE, Cheng M, Ashmun RA, Sherr CJ 1997 Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by
p16INK4a but not by the alternative reading frame protein p19ARF.
Proc Natl Acad Sci USA 94:669 – 673
65. Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A,
Alland L, Chin L, Potes J, Orlow I, Lee H-W, Cordon-Cardo C,
DePinho RA 1998 The Ink4a tumor suppressor gene product,
p19Arf, interacts with DM2 and neutralizes MDM29s inhibition of
p53. Cell 92:713–723
66. Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho
RA 1996 Role of the INK4a locus in tumor suppression and cell
mortality. Cell 85:27–37
67. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ 1997 Tumor suppression at the
mouse INK4a locus mediated by the alternative reading frame
product p19ARF. Cell 91:649 – 659
68. Zhang Y, Xiong Y, Yarbrough WG 1998 ARF promotes MDM2
degradation and stabilizes p53: ARF-INK4a locus deletion impairs
both the Rb and p53 tumor suppression pathways. Cell 92:725–734
69. Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes
S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G 1998 The
alternative product from the human CdkN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO
J 17:5001–5014
70. Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ
1998 Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA 95:8292– 8297
71. Sherr CJ 1998 Tumor surveillance via the ARF-p53 pathway. Genes
Dev 12:2984 –2991
72. El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R,
Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B 1993
WAF1, a potential mediator of p53 tumor suppression. Cell 75:
817– 825
73. Harper J, Adami GR, Wei N, Keyomarsi K, Elledge SJ 1993 The
p21 Cdk-interacting protein Cip1 is a potent inhibitor of the cyclin
D-dependent kinases. Cell 75:805– 816
74. Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM,
Tempst P, Massague J 1994 Cloning of p27Kip1, a cyclin-depen-
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
525
dent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78:59 – 66
Toyoshima H, Hunter T 1994 p27, A novel inhibitor of G1 cyclinCdk protein kinase activity, is related to p21. Cell 78:67–74
Lee M, Reynisdottir HI, Massague J 1995 Cloning of p57KIP2, a
cyclin-dependent kinase inhibitor with unique domain structure
and tissue distribution. Gene Dev 9:639 – 649
Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A,
Harper JW, Elledge SJ 1995 p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor
suppressor gene. Genes Dev 9:650 – 662
Watanabe H, Pan Z-Q, Schreiber-Agus N, DePinho RA, Hurwitz
J, Xiong Y 1998 Suppression of cell transformation by the cyclindependent kinase inhibitor p57KIP1 requires binding to proliferating cell nuclear antigen. Proc Natl Acad Sci USA 95:1392–1397
Hengst L, Gopfert U, Lashuel HA, Reed SI 1999 Complete inhibition of Cdk/cyclin by one molecule of p21Cip1. Genes Dev 12:
3882–3888
LaBaer J, Garret MD, Stevenson LF, Slingerland JM, Sandhu C,
Chou HS, Fattaey A, Harlow E 1997 New functional activities for
the p21 family of Cdk inhibitors. Genes Dev 11:847– 862
Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM,
Sherr CJ 1999 The p21Cip1 and p27Kip1 Cdk ’inhibitors’ are essential
activators of cyclin D-dependent kinases in murine fibroblasts.
EMBO J 18:1571–1583
Chen J, Kackson PK, Kirschner MW, Dutta A 1995 Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA.
Nature 374:386 –388
Goubin F, Ducommun B 1995 Identification of binding domains
on p21Cip1 cyclin-dependent kinase inhibitor. Oncogene 10:2281–
2287
Nakanishi M, Robetorye RS, Adams GR, Pereira-Smith OM,
Smith JR 1995 Identification of the active region of the DNA synthesis inhibitory gene p21Sdi1/CIP1/WAF1. EMBO J 14:555–563
Zhang H, Hannon GJ, Beach D 1994 p21-Containing cyclin kinases
exist in both active and inactive states. Genes Dev 8:1750 –1758
Harper JW, Elledge SJ, Keyomarski K, Dynlacht B, Tsai LH,
Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E
1995 Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell
6:387– 400
Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A 1996 Cyclinbinding motifs are essential for the function of p21CIP1. Mol Cell
Biol 16:4673– 4682
Luo Y, Hurwitz J, Massague J 1995 Cell-cycle inhibition by independent Cdk and PCNA binding domains in p21Cip1. Nature 375:
159 –161
Warbrick E, Lane D, Glover DM, Cox LS 1995 A small peptide
inhibitor of DNA replication defines the site of interaction between
the cyclin-dependent kinase inhibitor p21WAF1 and proliferating
cell nuclear antigen. Curr Biol 5:275–282
Flores-Rozas H, Kelman Z, Dean FB, Pan ZQ, Harper JW, Elledge
SJ, O’Donnell M, Hurwitz J 1994 Cdk-interacting protein 1 directly
binds with proliferating cell nuclear antigen and inhibits DNA
replication catalyzed by the DNA polymerase delta holoenzyme.
Proc Natl Acad Sci USA 91:8655– 8659
Deng C, Zhang P, Harper JW, Elledge SJ, Leder P 1995 Mice
lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82:675– 685
Reynisdottir I, Massague J 1997 The subcellular locations of
p15Ink4b and p27Kip1 coordinate their inhibitory interactions with
Cdk4 and Cdk2. Genes Dev 11:492–503
Coats S, Flanagan WM, Nourse J, Roberts JM 1996 Requirement
of p27Kip1 for restriction point control of the fibroblast cell cycle.
Science 272:877– 880
Rivard N, L’Allemain G, Bartek J, Pouyssegur J 1996 Abrogation
of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in
fibroblasts. J Biol Chem 271:18337–18341
Agrawal D, Dong KF, Wang YZ, Kayda D, Pledger WJ 1995
Regulation of cyclin E and p27Kip during mitosis in Balb/c3T3
cells. Cell Growth Differ 6:1199 –1205
Winston J, Dong F, Pledger WJ 1996 Differential modulation of G1
cyclins and the Cdk inhibitor p27kip1 by platelet-derived growth
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
526
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
PESTELL ET AL.
factor and plasma factors in density-arrested fibroblasts. J Biol
Chem 271:11253–11260
Firpo EJ, Koff A, Solomon E, Flanagan WM, Coats S, Polyak K,
Lee MH, Massague J, Crabtree GR, Roberts JM 1994 Inactivation
of a Cdk2 inhibitor during interleukin 2-induced proliferation of
human T lymphocytes. Mol Cell Biol 14:4889 – 4901
Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Sal GD,
Chau V, Yew PR, Draetta GF, Rolfe M 1995 Role of the ubiquitinproteosome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269:682– 685
Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts
JM, Koff A 1994 p27Kip1, a cyclin-Cdk inhibitor, links transforming
growth factor-b and contact inhibition to cell cycle arrest. Genes
Dev 8:9 –22
Kato J-Y, Matsuoka M, Strom DK, Sherr CJ 1994 Regulation of
cyclin D-dependent kinase 4 (Cdk4) by Cdk4-activating kinase. Mol
Cell Biol 14:2713–2721
Reynisdottir I, Polyak K, Iavarone A, Massague J 1995 Kip/Cip
and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in
response to TGF-b. Genes Dev 9:1831–1845
Soos T, Kiyokawa JH, Yan JS, Rubin MS, Giordano A, DeBlasio
A, Bottega S, Wong B, Mendelsohn J, Koff A 1996 Formation of
p27-Cdk complexes during the human mitotic cell cycle. Cell
Growth Differ 7:135–146
Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE 1997
Cyclin E-Cdk2 is a regulator of p27Kip1. Genes Dev 11:1464 –1478
Fero ML, Rivkin M, Tasch M, Peggy Porter P, Carow CE, Firpo
E, Polyak P, Tsai L-H, Broudy V, Perlmutter RM, Kaushansky K,
Roberts JM 1996 A syndrome of multiorgan hyperplasia with
features of gigantism, tumorigenesis, and female sterility in p27deficient mice. Cell 85:733–744
Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido
N, Horii IN, Loh DY, Nakayama K 1996 Mice lacking p27 display
increased body size, multiple organ hyperplasia, retinal dysplasia
and pituitary tumors. Cell 87:707–720
Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich V, Chao
MV, Koff A 1997 Oligodendrocyte precursor differentiation is
perturbed in the absence of the cyclin-dependent kinase inhibitor
p27Kip1. Genes Dev 11:2335–2346
Luo Y, Marx SO, Kiyokawa H, Koff A, Massague J, Marks AR
1996 Rapamycin resistance tied to defective regulation of p27Kip1.
Mol Cell Biol 16:6744 – 6751
Yan Y, Frisen J, Lee M-H, Massague J, Barbacid M 1997 Ablation
of the Cdk inhibitor p57Kip2 results in increased apoptosis and
delayed differentiation during mouse development. Genes Dev
11:973–983
Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson
JC, Silverman A, Harper JW, DePinho RA, Elledge SJ 1997 Altered
cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387:151–
158
Wang Y, Prives C 1995 Increased and altered DNA binding of p53
by S and G2/M but not G1 cyclin-dependent kinases. Nature 376:
88 –91
Lane S, Farlie P, Watson R 1997 B-Myb function can be markedly
enhanced by cyclin A-dependent kinase and protein truncation.
Oncogene 14:2445–2453
Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ 1997 Mitogen-activated and cyclin-dependent protein kinases selectively
and differentially modulate transcriptional enhancement by the
glucocorticoid receptor. Mol Cell Biol 17:3947–3954
Hara E, Hall M, Peters G 1997 Cdk2-dependent phosphorylation
of Id2 modulates activity of E2A-related transcription factors.
EMBO J 16:332–342
Zwijsen RML, Wientjens E, Klompmaker R, van der Sman J,
Bernards R, Michalides RJAM 1997 Cdk-independent activation
of estrogen receptor by cyclin D1. Cell 88:405– 415
Neuman E, Ladha M, Lin N, Upton TM, Miller SJ, DiRenzon J,
Pestell RG, Hinds PW, Dowdy SF, Brown M, Ewen ME 1997
Cyclin D1 stimulation of estrogen receptor transcription independent of Cdk4 activation. Mol Cell Biol 17:5338 –5347
Hirai H, Sherr C 1996 Interaction of D-type cyclins with a novel
myb-like transcription factor, DMP1. Mol Cell Biol 16:6457– 6467
Vol. 20, No. 4
117. Ganter B, Fu SI, Lipsick JS 1998 D-type cyclins repress transcriptional activation by the v-Myb but not the c-Myb DNA-binding
domain. EMBO J 17:255–268
118. Udvadia AJ, Rogers KT, Higgins PDR, Murata Y, Martin KH,
Humphrey PA, Horowitz JM 1993 Sp-1 binds promoter elements
regulated by the RB protein and Sp-1 mediated transcription is
stimulated by RB coexpression. Proc Natl Acad Sci USA 90:3265–
3269
119. Riley DJ, Lee E Y-HP, Lee WH 1994 The retinoblastoma protein,
more than a tumor suppressor. Annu Rev Cell Biol 10:1–29
120. Wang JY, Knudsen ES, Welch PJ 1994 The retinoblastoma tumor
suppressor protein. Adv Cancer Res 64:25– 85
121. Dynlacht B 1997 Regulation of transcription by proteins that control the cell cycle. Nature 389:149 –152
122. Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B,
Wessling HC, Morgan DO, Reinberg D 1995 Cdk-activating kinase complex is a component of human transcription factor TFIIH.
Nature 374:283–387
123. Serizawa H, Makela TP, Conaway JW, Conaway RC, Weinberg
RA, Young RA 1995 Association of Cdk-activating kinase subunits
with transcription factor TFIIH. Nature 374:280 –282
124. Rosignol M, Kolb-Cheynel I, Egly J-M 1997 Substrate specificity
of the Cdk-activating kinase (CAK) is altered upon association with
TFIIH. EMBO J 16:1628 –1637
125. Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ,
Kato J-K 1994 D-Type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol 14:2066 –2076
126. Yankulov KY, Bentley DL 1997 Regulation of Cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J 16:
1638 –1646
127. Peng J, Zhu Y, Proce DH 1998 Identification of multiple cyclin
subunits of human P-TEFb. Genes Dev 12:755–762
128. Gottesfeld JM, Wolf VJ, Dang T, Forbes DJ, Hartl P 1994 Mitotic
repression of RNA polymerase III transcription in vitro mediated
by phosphorylation of TFIIIB component. Science 263:81– 84
129. Segil N, Guermah M, Hoffmann A, Roeder RG, Heintz N 1996
Mitotic regulation of TFIID: inhibition of activator-dependent transcription and changes in subcellular localization. Genes Dev 10:
2389 –2400
130. Larminie C, Cairns CA, Mital R, Martin K, Kouzarides T, Jackson
SP, White RJ 1997 Mechanistic analysis of RNA polymerase III
regulation by the retinoblastoma protein. EMBO J 16:2061–2071
131. Martinez-Balbas M, Dey A, Rabindran S, Ozato K, Wu C 1995
Displacement of sequence specific transcription factors from mitotic chromatin. Cell 83:29 –38
132. Colgan DF, Murphy KGK, Prives C, Manley J 1996 Cell-cycle
regulation of poly(A) polymerase by phosphorylation. Nature 384:
282–285
133. Kitabayashi I, Eckner R, Arany Z, Chiu R, Gachelin G, Livingston
DM, Yokoyama KK 1995 Phosphorylation of the adenovirus E1Aassociated 300 kDa protein in response to retinoic acid and E1A
during the differentiation of F9 cells. EMBO J 14:3496 –3509
134. Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld
MG, Lazar MA 1996 A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol 16:5458 –5465
135. Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung
L 1996 Nuclear receptor coactivators and corepressors. Mol Endocrinol 1167–1177
136. Ogryzko VV, Schiltz RL, Russanova V, Howard B, Nakatani Y
1996 The transcriptional coactivators p300 and CBP are histone
acetyltransferases. Cell 87:953–959
137. Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty
CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E,
Eisenman RN, Rose DW, Glass CK, Rosenfeld MG 1997 A complex containing N-CoR, mSin3 and histone deacetylase mediates
transcriptional repression. Nature 387:43– 48
138. Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK,
Rosenfeld MG 1997 The transcriptional co-activator p/CIP binds
CBP and mediates nuclear-receptor function. Nature 387:677– 684
139. Missero C, Calautti E, Eckner R, Chin J, Tsai LH, Livingston DM,
Dotto GP 1995 Involvement of the cell-cycle inhibitor Cip/Waf1
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
and the E1A-associated p300 in terminal differentiation. Proc Natl
Acad Sci USA 92:5451–5455
Perkins ND, Felzian LK, Betts JC, Leung K, Beach D, Nabel GJ
1997 Regulation of NF-kB by cyclin-dependent kinases associated
with the p300 coactivator. Science 275:523–527
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine
AS, Kelly K 1997 Recruitment of p300/CBP in p53-dependent
signal pathways. Cell 89:1174 –1185
Stanley ER, Heard PM 1977 Factors regulating macrophage production and growth. Purification and some properties of the colony
stimulating factor from medium conditioned by mouse L cells.
J Biol Chem 252:4305– 4312
Stanley ER 1994 Colony stimulating factor-1 (macrophage colony
stimulating factor). In: Thompson A (ed) The Cytokine Handbook,
ed 2. Academic Press, Ltd., London, pp 387– 418
Hamilton JA 1997 CSF-1 signal transduction. J Leukoc Biol 62:
145–155
Hamilton JA 1997 CSF-1 signal transduction: what is of functional
significance? Trends Immunol Today 18:313–316
Buscher D, Sbarba PD, Hipskind RA, Rapp UR, Stanley ER,
Baccarini M 1993 v-raf Confers CSF-1 independent growth to a
macrophage cell line and leads to immediate early gene expression
without MAP-kinase activation. Oncogene 8:3323–3332
Langer SJ, Bortner DM, Roussel MF, Sherr CJ, Ostrowski MC
1992 Mitogenic signalling by colony-stimulating factor 1 and ras is
suppressed by the ets-2 DNA-binding domain and restored by myc
overexpression. Mol Cell Biol 12:5355–5362
Roussel MF, Theodoras AM, Pagano M, Sherr CJ 1995 Rescue of
defective mitogenic signaling by D-type cyclins. Proc Natl Acad Sci
USA 92:6837– 6841
Barone MV, Courtneidge SA 1995 Myc but not Fos rescue of PDGF
signalling block caused by kinase inactive Src. Nature 378:509 –511
Balk SD 1971 Calcium as a regulator of the proliferation of normal,
but not transformed, chicken fibroblasts in plasma-containing medium. Proc Natl Acad Sci USA 68:271–275
Olashaw NE, Olson JE, Drozdoff V, Pledger WJ 1992 Growth
factors: their role in the control of cell proliferation. In: Stein GS,
Lion JB (eds) Molecular and Cellular Approaches to the Control of
Proliferation and Differentiation. Academic Press, San Diego, CA,
pp 3–27
Stiles CD, Capone GT, Schwer CD, Antoniades HN, van Wyk JJ,
Pledger WJ 1979 Dual control of cell growth by somatomedins and
platelet derived growth factor. Proc Natl Acad Sci USA 76:1279 –
1283
Pardee AB 1989 G1 events and regulation of cell proliferation.
Science 256:603– 608
Won KA, Xiong Y, Beach D, Gilman MZ 1992 Growth-regulated
expression of D-type cyclin genes in human diploid fibloblasts.
Proc Natl Acad Sci USA 89:9910 –9914
Weber JD, Hu W, Jefcoat SC, Raben DM, Baldassare JJ 1997
Ras-stimulated extracellular signal-related kinase 1 and RhoA activities coordinate platelet-derived growth factor-induced G1 progression through the independent regulation of cyclin D1 and
p27KIP1. J Biol Chem 272:32966 –32971
Agrawal D, Hauser P, McPherson F, Dong F, Garcia A, Pledger
WJ 1996 Repression of p27kip1 synthesis by platelet derived growth
factor in BALB/c3T3 cells. Mol Cell Biol 16:4327– 4336
Armelin HA 1973 Pituitary extracts and steroid hormones in the
control of 3T3 cell growth. Proc Natl Acad Sci USA 70:2702–2706
Zhan X, Plourde X, Ku R, Maciag T 1994 Association of fibroblast
growth factor receptor-1 with c-src correlates with association between c-src and cortactin. J Biol Chem 269:20221–20224
Friesel RE, Maciag T 1995 Molecular mechanisms of angiogenesisfibroblast growth factor signal transduction. FASEB J 9:919 –925
Wang H, Rubin M, Fenig E, DeBlasio A, Mendelsohn J, Yahalom
J, Wieder R 1997 Basic fibroblast growth factor causes growth arrest
in MCF-7 human breast cancer cells while inducing both mitogenic
and inhibitory G1 events. Cancer Res 57:1750 –1757
Rao SS, Kohtz DS 1995 Positive and negative regulation of D-type
cyclin expression in skeletal myoblasts by basic fibroblast growth
factor and transforming growth factor b. A role for cyclin D1 in
control of myoblast differentiation. J Biol Chem 270:4093– 4100
Skapek S, Rhee J, Kim PS, Novitch B, Lassar A 1996 Cyclin
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
527
mediated inhibition of muscle gene expression via a mechanism
that is independent of pRB phosphorylation. Mol Cell Biol 16:
7043–7053
Skapek S, Rhee J, Spicer D, Lassar A 1995 Inhibition of myogenic
differentiation in proliferating myoblasts by cyclin D1-dependent
kinase. Science 267:1022–1024
Zhang J-M, Wei Q, Zhao X, Paterson BM 1999 Coupling of the cell
cycle and myogenesis through the cyclin-dependent interaction of
MyoD with Cdk4. EMBO J 18:926 –933
Leof EB, Van Wyk JJ, O’Keefe EJ, Pledger WJ 1983 Epidermal
growth factor (EGF) is required only during the traverse of early
G1 in PDGF stimulated density-arrested BALB/c-3T3 cells. Exp
Cell Res 147:202–208
Campisi J, Pardee AB 1984 Post transcriptional control of the onset
of DNA synthesis by an insulin-like growth factor. Mol Cell Biol
4:1807–1814
Olashaw NE, Van Wyk JJ, Pledger WJ 1987 Control of late G0/G1
progression and protein modification by SmC/IGF I. Am J Physiol
253:C575–C579
Jones JI, Clemmons DR 1995 Insulin-like growth factors and their
binding proteins: biological actions. Endocr Rev 16:3–34
Liu J-P, Baker J, Perkins AS, Robertson EJ, Efstratiadis A 1993
Mice containing null mutations of the genes encoding insulin-like
growth factor I (IGF-I) and type I IGF receptor (IGF-IR). Cell 75:
59 –72
Baker J, Liu J-P, Robertson EJ, Efstratiadis A 1993 Role of insulinlike growth factors in embryonic and postnatal growth. Cell 75:
73– 82
Sell C, Dumenil G, Deveaud C, Miura M, Coppola D, De Angelis
T, Rubin R, Efstratiadis A, Baserga R 1994 Effect of null mutation
of the type 1 IGF receptor gene on growth and transformation of
mouse embryo fibroblasts. Mol Cell Biol 14:3604 –3612
Vallentinis B, Porcu PL, Quinn K, Baserga R 1994 The role of the
insulin-like growth factor I receptor in the transformation by simian virus 40 T antigen. Oncogene 9:825– 831
Coppola D, Ferber A, Miura M, Sell C, D’Amrosio C, Rubin R,
Baserga R 1994 A functional insulin-like growth factor I receptor
is required for the mitogenic and transforming activiities of the
epidermal growth factor receptor. Mol Cell Biol 14:4588 – 4595
LeRoith D, Werner H, Beitner-Johnson D, Roberts CTJ 1995 Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev 16:143–163
Lowe WJ, Pestell RG, Madison L, Jameson JL 1998 Mechanisms
of hormone action. In: Jameson JL (ed) Principles of Molecular
Medicine. Blackwell Science, Massachusetts, pp 3–14
Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK,
Sutherland RL 1993 Growth factor, steroid, and steroid antagonist
regulation of cyclin gene expression associated with changes in
T-47D human breast cancer cell cycle progression. Mol Cell Biol
13:3577–3587
Sewing A, Burger C, Brusselbach S, Schalk C, Lucibello FC,
Muller R 1993 Human cyclin D1 encodes a labile nuclear protein
whose synthesis is directly induced by growth factors and suppressed by cyclic AMP. J Cell Sci 104:545–554
Furlanetto RW, Harwell SE, Frick KK 1994 Insulin-like growth
factor-I induces cyclin D1 expression in MG63 human osteosarcoma cells in vitro. Mol Endocrinol 8:510 –517
Shambaugh III GE, Lee RJ, Watanabe G, Erfurth F, Karnezis AN,
Koch AE, Haines III GK, Halloran M, Brodie BA, Pestell RG 1996
Reduced cyclin D1 expression in the cerebella of nutritionally deprived rats correlates with developmental delay and decreased
cellular DNA synthesis. J Neuropathol Exp Neurol 55:1010 –1021
Watanabe G, Pena P, Shambaugh III GE, Haines III GK, Pestell
RG 1998 Regulation of cyclin dependent kinase inhibitor proteins
during neonatal cerebellar development. Dev Brain Res 108:77– 87
Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C 1995 Mice
lacking cyclin D1 are small and show defects in eye and mammary
gland development. Genes Dev 9:2364 –2372
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H,
Haslam SZ, Bronson RT, Elledge SJ, Weinberg RA 1995 Cyclin D1
provides a link between development and oncogenesis in the retina
and breast. Cell 82:621– 630
Nourse J, Firpo E, Flanagan WM, Coats S, Polyak K, Lee MH,
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
528
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
PESTELL ET AL.
Massague J, Crabtree GR, Roberts JM 1994 Interleukin-2 mediated
elimination of the p27(KIP1) cyclin-dependent kinase inhibitor prevented by rapamycin. Nature 372:570 –573
Mann DJ, Higgins T, Jones NC, Rozengurt E 1997 Differential
control of cyclins D1 and D3 and the Cdk inhibitor p27Kip1 by
diverse signalling pathways in Swiss 3T3 cells. Oncogene 14:1759 –
1766
Earp HS, Dawson TL, Li X, Yu H 1995 Heterodimerization and
functional interaction between EGF receptor family members: a
new signaling paradigm with implications for breast cancer research. Breast Cancer Res Treat 35:115–132
Carpenter G, Wahl MI 1990 The epidermal growth factor family.
In: Sporn MB, Roberts AB (eds) Handbook of Experimental Pharmacology, Peptide Growth Factors, and Their Receptors. SpringerVerlag, Berlin, pp 69 –171
Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A,
Pestell RG 1995 Transforming p21ras mutants and c-Ets-2 activate
the cyclin D1 promoter through distinguishable regions. J Biol
Chem 270:23589 –23597
Yan Y-X, Nakagawa H, Lee M-H, Rustgi AK 1997 Transforming
growth factor-a enhances cyclin D1 transcription through the binding of early growth factor response protein to a cis-regulatory
element in the cyclin D1 promoter. J Biol Chem 272:33181–33190
Deleted in proof
Mark MD, Storm DR 1997 Coupling of epidermal growth factor
(EGF) with the antiproliferative activity of cAMP induces neuronal
differentiation. J Biol Chem 272:17238 –17244
Takuwa N, Takuwa Y 1997 Ras activity late in G1 phase required
for p27 kip1 downregulation, passage through the restriction point,
and entry into S phase in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol 17:5348 –5358
Barbacid M 1993 Nerve growth factor: a tale of two receptors.
Oncogene 8:2033–2042
Ginty DD, Bonni A, Greenberg ME 1994 Nerve growth factor
activates a ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77:713–725
Pumiglia KM, Decker SJ 1997 Cell cycle arrest mediated by the
MEK/mitogen-activated protein kinase pathway. Proc Natl Acad
Sci USA 94:448 – 452
Buchkovich KJ, Ziff EB 1994 Nerve growth factor regulates the
expression and activity of p33Cdk2 and p34cdc2 kinases in PC12
pheochromocytoma cells. Mol Biol Cell 5:1225–1241
Yan G-Z, Ziff EB 1995 NGF regulates the PC12 cell cycle machinery
through specific inhibition of the Cdk kinases and induction of
cyclin D1. J Neurosci 15:6200 – 6212
van Grunsven LA, Thomas A, Urdailes JL, Machenaud S, Choler
P, Durnad I, Rudkin BB 1996 Nerve growth factor-induced accumulation of PC12 cells expressing cyclin D1: evidence for a G1
phase block. Oncogene 12:855– 862
Yan G, Ziff EB 1997 Nerve growth factor induces transcription of
the p21 WAF1/CIP1 and cyclin D1 genes in PC12 cells by activating
the Sp1 transcription factor. J Neurosci 17:6122– 6132
Park DS, Farinelli SE, Greene LA 1996 Inhibitors of cyclindependent kinases promote survival of post-mitotic neuronally
differentiated PC12 cells and sympathetic neurons. J Biol Chem
271:8161– 8169
Kranenburg O, van der Eb AJ, Zantema A 1996 Cyclin D1 is an
essential mediator of apoptotic neuronal cell death. EMBO J 15:
46 –54
Han EK, Begemann M, Sgambato A, Soh JW, Doki Y, Xing WQ,
Liu W, Weinstein IB 1996 Increased expression of cyclin D1 in a
murine mammary epithelial cell line induces p27 kip1, inhibits
growth and enhances apoptosis. Cell Growth Differ 7:699 –710
Sofer-Levi Y, Resnitzky D 1996 Apoptosis induced by ectopic
expression of cyclin D1 but not cyclin E. Oncogene 13:2431–2437
Pardo FS, Su M, Borek C 1996 Cyclin D1 induced apoptosis maintains the integrity of the G1/S checkpoint following ionizing radiation irradiation. Somat Cell Mol Genet 22:135–144
Del Sal G, Murphy M, Ruaro EM, Lazarevic D, Levine AJ, Schneider C 1996 Cyclin D1 and p21/waf1 are both involved in p53
growth suppression. Oncogene 12:177–185
Nikolic M, Dudek H, Kwon YT, Ramos YFM, Tsai L-H 1996 The
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
229.
Vol. 20, No. 4
Cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10:816 – 825
Movsesyan V, Whalin M, Shibutani M, Katagiri Y, Broude E,
Guroff G 1996 Down-regulation of cyclin F levels during nerve
growth factor-induced differentiation of PC12 cells. Exp Cell Res
227:203–207
Roberts AB, Sporn MB 1990 The transforming growth factor-bs. In:
Sporn MB, Roberts AB (eds) Peptide Growth Factors and Their
Receptors. Springer-Verlag, Berlin, pp 419 – 472
Zhao Y, Young SL 1996 Requirement of transforming growth factor-b (TGF-b) type II receptor for TGF-b-induced proliferation and
growth inhibition. J Biol Chem 271:2369 –2372
Kay EP, Lee HK, Park KS, Lee SC 1998 Indirect mitogenic effect
of transforming growth factor-b on cell proliferation of subconjunctival fibroblasts. Invest Opthalmol Vis Sci 39:481– 486
Grotendorst GR 1997 Connective tissue growth factor: a mediator
of TGF-b action on fibroblasts. Cytokine Growth Factor Rev 8:171–
179
Alexandrov MG, Moses HL 1995 Transforming growth factor b
and cell cycle regulation. Cancer Res 55:1452–1457
Markowitz SD, Roberts AB 1996 Tumor suppressor activity of the
TGFb pathway in human cancers. Cytokine Growth Factor Rev
1:93–102
Bottinger EP, Letterio JJ, Roberts AB 1997 Biology of TGFb in
knockout and transgenic mouse models. Kidney Int 51:1355–1360
Penton A, Selleck SB, Hoffmann FM 1997 Regulation of cell cycle
synchronization by decapentaplegic during Drosophila eye development. Science 275:203–206
Derynck R, Zhang Y 1996 Intracellular signaling: the mad way to
do it. Curr Biol 6:1226 –1269
Zhang Y, Musci T, Derynck R 1997 The tumor suppressor Smad4/
DPC 4 as a central mediator of Smad function. Curr Biol 7:270 –276
Zhang Y, Feng X, We R, Derynck R 1996 Receptor-associated Mad
homologues synergize as effectors of the TGF-b response. Nature
383:168 –172
Wu RY, Zhang Y, Feng XH, Derynck R 1997 Heteromeric and
homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol Cell Biol
17:2521–2528
Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein
B, Kern SE 1998 Human Smad3 and Smad4 are sequence-specific
transcription activators. Molecular Cell 1:611– 617
Howe PH, Draetta G, Leof EB 1991 Transforming growth factor b1
inhibition of p34cdc2 phosphorylation and histone H1 kinase activity is associated with a G1/S-phase growth arrest. Mol Cell Biol
11:1185–1194
Wu F, Buckley S, Bui KC, Yee A, Wu HY, Liu J, Warburton D 1996
Cell cycle arrest in G0/G1 phase by contact inhibition and TGF-b
1 in mink Mv1Lu lung epithelial cells. Am J Physiol 270:L879 –L888
Ewen ME, Sluss HK, Whitehouse LL, Livingston DM 1993 TGFb
inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell
74:1009 –1020
Sandhu C, Garbe J, Bhattacharya N, Daksis J, Pan CH, Yaswen P,
Koh J, Slingerland JM, Stampfer MR 1997 Transforming growth
factor b stabilizes p15INK4B protein, increases p15INK4b-Cdk4 complexes, and inhibits cyclin D1-Cdk4 association in human mammary epithelial cells. Mol Cell Biol 17:2458 –2467
Florenes VA, Bhattacharya N, Bani MR, Ben-David Y, Kerbel RS,
Slingerland JM 1996 TGF-b mediated G1 arrest in a human melanoma cell line lacking p15INK4b. Evidence for cooperation between
p21Cip1/WAF1 and p27Kip1. Oncogene 13:2447–2457
Iavarone A, Massague J 1997 Repression of the Cdk activator
Cdc25A and cell-cycle arrest by cytokine TGF-b in cells lacking the
Cdk inhibitor p15. Nature 387:417– 422
Herrera RE, Makela TP, Weinberg RA 1996 TGFb-induced growth
inhibition in primary fibroblasts requires the retinoblastoma protein. Mol Biol Cell 7:1335–1342
Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche
D 1998 The three members of the pocket proteins family share the
ability to repress E2F activity through recruitment of a histone
deacetylase. Proc Natl Acad Sci USA 95:10493–10498
Deleted in proof
Eto Y, Tsui T, Takezawa M, Takano Y, Yokogawa Y, Shibai H 1987
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
250.
251.
252.
253.
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
Purification and characterization of erythroid differentiation factor
(EDF) isolated from human leukemia cell line THP-1. Biochem
Biophys Res Commun 142:1095–1103
Sokol S, Melton DA 1991 Pre-existent patterns in Xenopus pole
cells revealed by induction with activin. Nature 351:409 – 411
Smith JC 1993 Mesoderm-inducing factors in early development.
EMBO J 12:4463– 4470
Wang QF, Tilly KI, Tilly JL, Preffer F, Schneyer AL, Crowley Jr
WF, Sluss PM 1996 Activin inhibits basal and androgen-stimulated
proliferation and induces apoptosis in the human prostatic cancer
cell line, LNCaP. Endocrinology 137:5476 –5483
Yamato K, Koseki T, Ohguchi M, Kiazaki M, Ikeda Y, Nishihara
T 1997 Activin A induction of cell-cycle arrest involves modulation
of cyclin D2 and p21CIP/WAF1 in plasmacytic cells. Mol Endocrinol
11:1044 –1052
Natarajan R, Gonzales N, Hornsby PJ, Nadler J 1992 Mechanism
of angiotensin II-induced proliferation in bovine adrenocortical
cells. Endocrinology 131:1174 –1180
Clyne CD, Nicol MR, MacDonald S, Williams BC, Walker SW
1993 Angiotensin II stimulates growth and steroidogenesis in zona
fasciculata/reticularis cells from bovine adrenal cortex via the AT1
receptor subtype. Endocrinology 132:2206 –2212
Sadoshima J, Izumo S 1993 Signal transduction pathways of angiotensin II-induced c-fos gene expression in cardiac myocytes in
vitro. Roles of phospholipid-derived second messengers. Circ Res
73:413– 423
Weber H, Taylor DS, Molloy CJ 1994 Angiotensin II induces
delayed mitogenesis and cellular proliferation in rat aortic smooth
muscle cells. J Clin Invest 93:788 –798
Campbell-Boswell M, Robertson AL 1981 Effects of angiotensin II
and vasopressin on human smooth muscle cells in vitro. Exp Mol
Pathol 35:265–276
Neer EJ 1995 Heterotrimeric G proteins: organizers of transmembrane signals. Cell 80:249 –257
Clapham DE 1995 Calcium signaling. Cell 80:259 –268
Barrett PQ, Bollag WB, Isales CM, McCarthy RT, Rasmussen H
1989 Role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev 10:495–518
Chao TS, Foster DA, Rapp ER, Rosner MR 1994 Differential Raf
requirement for activation of mitogen-activated protein kinase by
growth factors, phorbol esters and calcium. J Biol Chem 269:7337–7341
Chao T-SO, Byron KL, Lee K-M, Villereal M, Rosner MR 1992
Activation of MAP kinases by calcium-dependent and calciumindependent pathways. Stimulation by thapsigargin and epidermal growth factor. J Biol Chem 267:19876 –19883
Lin L-L, Wartman M, Lin AY, Knopf JL, Seth A, Davis RJ 1993 cPLA2
is phosphorylated and activated by MAP kinase. Cell 72:269–278
Ghosh A, Greenberg ME 1995 Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268:239–246
Schorb W, Peeler TC, Madigan NN, Conrad KM, Baker KM 1994
Angiotensin II-induced protein phosphorylation in neonatal rat
cardiac fibroblasts. J Biol Chem 269:19626 –19632
Marrero MB, Paxton WG, Duff JL, Berk BC, Bernstein KE 1994
Angiotensin II stimulates tyrosine phosphorylation of phospholipase C-gamma 1 in vascular smooth muscle cells. J Biol Chem
269:10935–10939
Zohn IE, Xiong HY, Cox AD, Earp HS 1995 Angiotensin II stimulates calcium dependent activation of c-Jun N-terminal kinase.
Mol Cell Biol 15:6160 – 6168
Marrero MB, Scheiffer B, Paxton WG, Heerdt L, Berk BC,
Delafontaine P, Bernstein KE 1995 Direct stimulation of Jak/STAT
pathway by the angiotensin II AT1 receptor. Nature 375:247–250
Watanabe G, Lee RJ, Albanese C, Rainey WE, Batlle D, Pestell RG
1996 Angiotensin II (AII) activation of cyclin D1-dependent kinase
activity. J Biol Chem 271:22570 –22577
Onishi T, Hruska K 1997 Expression of p27Kip1 in osteoblast-like
cells during differentiation with parathyroid hormone. Endocrinology 138:1995–2004
Bianchi S, Fabiani S, Muratori M, Arnold A, Sakaguchi K, Miki
T, Brandi ML 1994 Calcium modulates the cyclin D1 expression in
a rat parathyroid cell line. Biochem Biophys Res Commun 204:
691–700
Dumont JE, Lamy F, Roger P, Maenhaut C 1992 Physiological and
254.
255.
256.
257.
258.
259.
260.
261.
262.
263.
264.
265.
266.
267.
268.
269.
270.
271.
272.
273.
274.
529
pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiol Rev 72:667– 697
Tramontano D, Chin WW, Moses AC, Ingbar SH 1986 Thyrotropin and dibutyryl cyclic AMP increase levels of c-myc and c-fos
mRNAs in cultured rat thyroid cells. J Biol Chem 261:3919 –3922
Woloshin PI, Walton KM, Rehfuss RP, Goodman RH, Cone RD
1992 39,59-Cyclic adenosine monophosphate-regulated enhancer
binding (CREB) activity is required for normal growth and differentiated phenotype in the FRTL5 thyroid follicular cell line. Mol
Endocrinol 6:1725–1733
Yamamoto K, Hirai A, Ban T, Saito J, Tahara K, Terano T, Tamura
Y, Saito Y, Kitagawa M 1996 Thyrotropin induces G1 cyclin expression and accelerates G1 phase after insulin-like growth factor
I stimulation in FRTL-5 cells. Endocrinology 137:2036 –2042
Lazzereschi D, Coppa A, Minicione G, Lavitrano M, Fragomele
F, Colletta G 1997 The phosphatase inhibitor okadaic acid stimulates the TSH-induced G1-S phase transition in thyroid cells. Exp
Cell Res 234:425– 433
Depoortere F, Van Keymeulen A, Lukas J, Costagliola S, Bartkova
J, Dumont JE, Bartek J, Roger PP, Dremier S 1998 A requirement
for cyclin D3-cyclin-dependent kinase (Cdk)-4 assembly in the cyclic adenosine monophosphate-dependent proliferation of thyrocytes. J Cell Biol 140:1427–1439
Richards JS 1994 Hormonal control of gene expression in the ovary.
Endocr Rev 15:725–751
Desjardins C, Ewing LL 1993 Cell and Molecular Biology of the
Testis. Oxford University Press, New York
Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY,
Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ,
Bronson RT, Elledge SJ, Weinberg RA 1996 Cyclin D2 is an FSHresponsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384:470 – 474
Robker RL, Richards JS 1998 Hormone-induced proliferation and
differentiation of granulosa cells: a coordinated balance of the cell
cycle regulators cyclin D2 and p27Kip1. Mol Endocrinol 12:924 –940
Dickson RB, Lippman ME 1995 Growth factors in breast cancer.
Endocr Rev 16:559 –589
Sunderland MC, McGuire WL 1991 Oncogenes as clinical prognostic indicators. In: Lippman M, Dickson R (eds) Regulatory
Mechanisms in Breast Cancer. Kluwer Academic Publishers, Boston, MA, pp 3–22
Musgrove EA, Sutherland RL 1991 Steroids, growth factors and
cell cycle controls in breast cancer. Cancer Treat Res 53:305–331
Davis VL, Couse JF, Goulding EH, Power SG, Eddy EM, Korach
KS 1994 Aberrant reproductive phenotypes evident in transgenic
mice expressing the wild-type mouse estrogen receptor. Endocrinology 135:379 –386
Korach KS, Couse JF, Curtis SW, Washburn TF, Lindzy J, Kimbro
KS, Eddy EM, Migliaccio S, Snedeker SM, Lubahn DB,
Schomberg DW, Smith EP 1996 Estrogen receptor gene disruption:
molecular characterization and experimental and clinical phenotypes. Recent Prog Horm Res 51:159 –186
Johns A, Freay AD, Fraser W, Korach KS, Rubanyi GM 1996 Disruption of estrogen receptor gene prevents 17b-estradiol-induced angiogenesis in transgenic mice. Endocrinology 137:4511– 4523
Eddy EM, Washburn TF, Bunch DO, Goulding EH, Gladen BC,
Lubahn DB, Korach KS 1996 Targeted disruption of the estrogen
receptor gene in male mice causes alteration of spermatogenesis
and infertility. Endocrinology 137:4796 – 4805
Beato M 1989 Gene regulation by steroid hormones. Cell 56:335–
344
Truss M, Beato M 1993 Steroid hormone receptors: interaction with
deoxyribonucleic acid and transcription factors. Endocr Rev 14:
459 – 479
Migliaccio A, DiDomenico M, Castoria G, deFalco A, Bontempo
P, Nola E, Auricchio F 1996 Tyrosine kinase/p21ras/MAP-kinase
pathway activation by estradiol-receptor complex in MCF-7 cells.
EMBO J 15:1292–1300
Ignar-Trowbridge DM, Pimentel M, Parker MG, McLachlan JA,
Korach KS 1996 Peptide growth factor cross-talk with the estrogen
receptor requires the A/B domain and occurs independently of
protein kinase C or estradiol. Endocrinology 137:1735–1744
Schuchard M, Landers JP, Sandhu NP, Spelsberg TC 1993 Steroid
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
530
275.
276.
277.
278.
279.
280.
281.
282.
283.
284.
285.
286.
287.
288.
289.
290.
291.
292.
293.
294.
295.
PESTELL ET AL.
hormone regulation of nuclear proto-oncogenes. Endocr Rev 14:
659 – 669
Sumi-Ichinose C, Ichinose H, Metzger D, Chambon P 1997
SNF2b-BRG1 is essential for the viability of F9 murine embryonal
carcinoma cells. Mol Cell Biol 17:5976 –5986
Mengus G, May M, Carre L, Chambon P, Davidson I 1997 Human
TAF(II)135 potentiates transcriptional activation by the AF-2s of the
retinoic acid, vitamin D3 and thyroid hormone receptors in mammalian cells. Genes Dev 11:1381–1395
Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin
F, Losson R, Chambon P 1996 A possible involvement of TIF1 a
and TIF1 b in the epigenetic control of transcription by nuclear
receptors. EMBO J 15:6701– 6715
O’Malley BW 1997 Role of co-activators and co-repressors in the
mechanism of steroid/thyroid receptor action. Recent Prog Horm
Res 52:141–164
Lippman M, Bolan G, Huff K 1976 The effects of estrogens and
antiestrogens on hormone responsive human breast cancer in long
term tissue culture. Cancer Res 36:4595– 4601
Lippman M, Bolan G 1975 Oestrogen-responsive human breast
cancer in long term tissue culture. Nature 256:592–593
Leung BS, Potter AH 1987 Mode of estrogen action on cell proliferation in CAMA-1 cells. II. Sensitivity of G1 phase population.
J Cell Biochem 34:213–225
Foster JS, Wimalasena J 1996 Estrogen regulates activity of cyclindependent kinases and retinoblastoma protein phosphorylation in
breast cancer cells. Mol Endocrinol 10:488 – 498
Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M,
Beato M, Sica V, Bresciani F, Weisz A 1996 17b-Estradiol induces
cyclin D1 gene transcription, p36D1–p34Cdk4 complex activation
and p105Rb phosphorylation during mitogenic stimulation of G(1)arrested human breast cancer cells. Oncogene 12:2315–2324
Musgrove EA, Sarcevic B, Sutherland RL 1996 Inducible expression of cyclin D1 in T-47D human breast cancer cells is sufficient
for Cdk2 activation and pRB hyperphosphorylation. J Cell Biochem
60:363–378
Bunone G, Briand P-A, Miksicek RJ, Picard D 1996 Activation of
the unliganded estrogen receptor by EGF involves the MAP kinase
pathway and direct phosphorylation. EMBO J 15:2174 –2183
Planas-Silva MD, Weinberg RA 1997 Estrogen-dependent cyclin
E-Cdk2 activation through p21 redistribution. Mol Cell Biol 17:
4059 – 4069
Prall OWJ, Sarcevic B, Musgrove EA, Watts CKW, Sutherland RL
1997 Estrogen-induced activation of Cdk4 and Cdk2 during G1-S
phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association
with cyclin E-Cdk2. J Biol Chem 272:10882–10894
Prall OWJ, Rogan EM, Musgrove EA, Watts CKW, Sutherland RL
1998 c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2
activation and cell cycle reentry. Mol Cell Biol 18:4499 – 4508
Cunha GR, Young P, Hom YK, Cooke PS, Taylor JA, Lubahn DB
1997 Elucidation of a role for stromal steroid hormone receptors in
mammary gland growth and development using tissue recombinants. J Mamm Gland Biol Neoplasia 2:393– 402
Zwijsen RML, Buckle RS, Hijmans EM, Loomans CJM, Bernards
R 1998 Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev 12:3488 –3498
Heery DM, Kalkhoven E, Hoare S, Parker MG 1997 A signature
motif in transcriptional co-activators mediates binding to nuclear
receptors. Nature 387:733–736
Trowbridge JM, Rogatsky I, Garabedian MJ 1997 Regulation of
estrogen receptor transcriptional enhancement by the cyclin
A/Cdk2 complex. Proc Natl Acad Sci USA 94:10132–10137
Couse JF, Lindzey J, Grandien K, Gustafsson JA, Korach KS 1997
Tissue distribution and quantitative analysis of estrogen receptor-a
(ERa) and estrogen receptor-b (ERb) messenger ribonucleic acid in the
wild-type and ERa-knockout mouse. Endocrinology 138:4613–4621
Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner
PJ, Scanlan TS 1997 Differential ligand activation of estrogen receptors ERa and ERb at AP1 sites. Science 277:1508 –1510
Altucci L, Addeo R, Cicatiello L, Germano D, Pacilio C, Battista
T, Cancemi M, Petrizzi VB, Bresciani F, Weisz A 1997 Estrogen
induces early and timed activation of cyclin-dependent kinases 4,
296.
297.
298.
299.
300.
301.
302.
303.
304.
305.
306.
307.
308.
309.
310.
311.
312.
313.
314.
315.
Vol. 20, No. 4
5, and 6 and increases cyclin messenger ribonucleic acid expression
in rat uterus. Endocrinology 138:978 –984
Bresciani F 1968 Topography of DNA synthesis in the mammary
gland of the C3H mouse and its control by ovarian hormones. An
autoradiographic study. Cell Tissue Kinetics 1:51– 63
Shyamala G 1987 Endocrine and other influences in the normal
development of the breast. In: Paterson AHG, Lees AW (eds) Fundamental Problems in Breast Cancer. Marinus Nijoff, Boston, pp
127–137
Michna H, Schneider MR, Nishino Y, El Etreby MF 1989 Antitumor activity of the antiprogestins ZK 98.299 and RU 38.486 in
hormone dependent rat and mouse mammary tumors. Mechanistic
studies. Breast Cancer Res Treat 14:275–288
Shyamala G 1997 Roles of estrogen and Progesterone in normal
mammary gland development. Trends Endocrinol Metab 8:34 –39
Lydon JP, DeMayo J, Funk CR, Mani SK, Hughes AR, Montgomery CA, Shyamala G, Conneely OM, O’Malley BW 1995 Mice
lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev 9:2266 –2278
Musgrove EA, Lee CS, Sutherland RL 1991 Progestins both stimulate and inhibit breast cancer cell cycle progression while increasing expression of transforming growth factor a, epidermal growth
factor receptor, c-fos, and c-myc genes. Mol Cell Biol 11:5032–5043
Horwitz KB 1992 The molecular biology of RU486. Is there a role
for antiprogestins in the treatment of breast cancer? Endocr Rev
13:146 –163
Musgrove EA, Lee CSL, Cornish AL, Swarbrick A, Sutherland RL
1997 Antiprogestin inhibition of cell cycle progression in T-47D
breast cancer cells is accompanied by induction of the cyclindependent kinase inhibitor p21. Mol Endocrinol 11:54 – 66
Said TK, Conneely OM, Medina D, O’Malley BW, Lydon JP 1997
Progesterone, in addition to estrogen, induces cyclin D1 expression
in the murine mammary epithelial cell, in vivo. Endocrinology
138:3933–3939
Gudas LJ, Sporn MB, Roberts AB 1994 Cellular biology and biochemistry of the retinoids. In: Sporn MB, Roberts AB, Goodman DS
(eds) The Retinoids, ed 2. Raven Press, New York, pp 443–520
Mader S, Chen JY, Chen Z, White J, Chambon P, Gronemeyer H
1993 The patterns of binding of RAR, RXR and TR homo- and
heterodimers to direct repeats are dictated by the binding specificities of the DNA binding domains. EMBO J 12:5029 –5041
Forman B, Umesono K, Chen J, Evans R 1995 Unique response
pathways are established by allosteric interactions among nuclear
hormone receptors. Cell 81:541–550
Kim KH, Stellmack V, Javors J, Fuchs E 1987 Regulation of human
mesothelial cell differentiation: opposing roles of retinoids and
epidermal growth factor in the expression of intermediate filament
proteins. J Cell Biol 105:3039 –3051
Hudson LG, Santon JB, Glass CK, Gill GN 1990 Ligand-activated
thyroid hormone and retinoic acid receptors inhibit growth factor
receptor promoter expression. Cell 62:1165–1175
Liu M, Iavarone A, Freedman LP 1996 Transcriptional activation
of the human p21(WAF1/CIP1) gene by retinoic acid receptor.
Correlation with retinoid induction of U937 cell differentiation.
J Biol Chem 271:31723–31728
Langenfeld J, Kiyokawa H, Sekula D, Boyle J, Dmitrovsky E 1997
Posttranslational regulation of cyclin D1 by retinoic acid: a chemoprevention mechanism. Proc Natl Acad Sci USA 94:12070 –12074
Costa SL, Pratt C, McBurney MW 1996 E2F inhibits transcriptional
activation by the retinoic acid receptor. Cell Growth Differ 7:1479 –
1485
Yang-Yen H-f Zhang XK, Graupner G, Tzukerman M, Sakamoto
B, Karin M, Pfahl M 1991 Antagonism between retinoic acid receptors and AP-1: implications for tumor promotion and inflammation. New Biol 3:1206 –1219
Pestell RG, Jameson JL 1995 Hormone action II: transcriptional
regulation of endocrine genes by second messenger signalling
pathways. In: Weintraub B (ed) Molecular Endocrinology: Basic
Concepts and Clinical Correlations. Raven Press, New York, chapter 5, pp 59 –76
Chen J-Y, Penco S, Ostrowski J, Balaguer P, Pons M, Chambon
P, Gronenmeyer H 1995 RAR-specific agonist/antagonists which
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
316.
317.
318.
319.
320.
321.
322.
323.
324.
325.
326.
327.
328.
329.
330.
331.
332.
333.
334.
335.
336.
337.
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
dissociate transactivation and AP1 transrepression inhibit anchorage-independent cell proliferation. EMBO J 14:1187–1197
Kamei Y, Xu L, Heinzel T, Torchia J, Kurakawa R, Gloss B, Lin
S-C, Heyman RA, Rose DW, Glass CK, Rosenfeld MG 1996 A CBP
integrator complex mediates transcriptional activation and AP-1
inhibition by nuclear receptors. Cell 85:403– 414
Glass CK, Rose DW, Rosenfeld MG 1997 Nuclear receptor coactivators. Curr Opin Cell Biol 9:222–232
Bamberger CH, Schulte H, Chrousos GP 1996 Molecular determinants of glucocorticoid receptor function and tissue sensitivity
to glucocorticoids. Endocr Rev 17:245–261
Martin DJ, Tomkins GM, Granner D 1969 Synthesis and induction
of tyrosine aminotransferase in synchronized hepatoma cells in
culture. Proc Natl Acad Sci USA 62:248 –255
Hsu SC, Qi M, DeFranco DB 1992 Cell cycle regulation of glucocorticoid receptor function. EMBO J 11:3457–3468
Fanger BO, Currie RA, Cidlowski JA 1986 Regulation of epidermal
growth factor receptors by glucocorticoids during the cell cycle in
HeLa S3 cells. Arch Biochem Biophys 249:116 –125
Currie RA, Cidlowski JA 1982 Identification of modified forms of
human glucocorticoid receptors during the cell cycle. Endocrinology 110:2192–2194
Orti E, Mendel DB, Smith LL, Munck A 1989 Agonist dependent
phosphorylation and nuclear dephosphorylation of glucocorticoid
receptors in intact cells. J Biol Chem 264:9728 –9731
Hu JM, Bodwell JE, Munck A 1994 Cell cycle-dependent glucocorticoid receptor phosphorylation and activity. Mol Endocrinol
8:2609 –2616
Mason SA, DeFranco DB 1993 Site directed mutagenesis of the
phosphorylation sites of the mouse glucocorticoid receptor. J Biol
Chem 268:21501–21504
Almlof T, Wright AP, Gustafsson J-A 1995 Role of acidic and
phosphorylated residues in gene activation by the glucocorticoid
receptor. J Biol Chem 270:17535–17540
Orti E, Hu LM, Munck A 1993 Kinetics of glucocorticoid receptor
phosphorylation in intact cells. Evidence for hormone-induced hyperphosphorylation after activation and recycling of hyperphosphorylated receptors. J Biol Chem 268:7779 –7784
Ramalingam A, Hirai A, Thompson EA 1997 Inhibition of fibroblast proliferation and regulation of the cyclin kinase inhibitor
p21Cip1. Mol Endocrinol 11:577–586
Corroyer S, Nabeyrat E, Clement A 1997 Involvement of the cell
cycle inhibitor CIP1/WAF1 in lung alveolar epithelial cell growth
arrest induced by glucocorticoids. Endocrinology 138:3677–3685
Rogatsky I, Trowbridge JM, Garabedian MJ 1997 Glucocorticoid
receptor-mediated cell cycle arrest is achieved through distinct
cell-specific transcriptional regulatory mechanisms. Mol Cell Biol
17:3181–3193
Scheinman RI, Cogswell PC, Lofquist AK, Baldwin Jr AS 1995
Role of transcriptional activation of IkBa in mediation of immunosuppression by glucocorticoids. Science 270:283–286
Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M 1995
Immunosuppression by glucocorticoids:Inhibition of NF-kB activity through induction of IkB synthesis. Science 270:286 –290
Bouillon R, Okamura WH, Norman AW 1995 Structure-function
relationships in the vitamin D endocrine system. Endocr Rev 16:
200 –257
Frampton RJ, Omond SA, Eisman JA 1983 Inhibition of human
cancer cell growth by 1,25-dihydroxyvitamin D3 metabolites. Cancer Res 43:4443– 4447
Gross M, Kost SB, Ennis B, Stumpf W, Kumar R 1986 Effect of
1,25-dihydroxyvitamin D3 on mouse mammary tumor (GR) cells.
Evidence for receptors, cellular uptake, inhibition of growth and
alteration in morphology at physiologic concentrations of hormone. J Bone Miner Res 1:457– 467
Elstner E, Linker-Israeli M, Said J, Umiel T, de Vos S, Shintaku
I, Heber D, Binderup L, Uskokovic M, Koeffler HP 1995 20-epivitamin D3 analogues: a novel class of potent inhibitors of proliferation and inducers of differentiation of human breast cancer cell
lines. Cancer Res 55:2822–2830
Oikawa T, Yoshida Y, Shimamura M, Ashino-Fuse H, Iwaguchi
T, Tominaga T 1991 Antitumor effect of 22-oxa-1a,25-dihydroxyvitamin D3, a potent angiogenesis inhibitor, on rat mammary tumors
338.
339.
340.
341.
342.
343.
344.
345.
346.
347.
348.
349.
350.
351.
352.
353.
354.
355.
356.
357.
358.
531
induced by 7,12-dimethylbenz[a]anthracene. Anticancer Drugs
2:475– 480
Colston KW, Chander SK, Mackay AG, Coombes RC 1992 Effects
of synthetic vitamin D analogues on breast cancer cell proliferation
in vivo and in vitro. Biochem Pharmacol 44:693–702
Saez S, Falette N, Guillot C, Meggouh F, Lefebvre MF, Crepin M
1993 1,25(OH)2D3 modulation of mammary tumor cell growth in
vitro and in vivo. Breast Cancer Res 27:69 – 81
Bikle DD, Pillai S 1993 Vitamin D, calcium and epidermal differentiation. Endocr Rev 14:3–19
Moromoto S, Imanaka S, Koh E, Shiraishi T, Nabata T, Kitano S,
Miyashita Y, Nishii Y, Ogihara T 1989 Comparisons of the inhibitions of proliferation of normal and psoriatic fibroblasts by 1a,25dihydroxyvitamin D3 and synthetic analogues of vitamin D3 with
an oxygen atom in their side chain. Biochem Int 19:1143–1149
de Vos S, Holden S, Heber D, Elstner E, Binderup L, Uskokovic
M, Rude B, Chen DL, Le J, Cho SK, Koeffler HP 1997 Effects of
potent vitamin D3 analogs on clonal proliferation of human prostate cancer cell lines. Prostate 31:77– 83
Tang W, Ziboh VA, Isseroff R, Martinez D 1987 Novel regulatory
action of 1,25-dihydroxyvitamin D on the metabolism of polyphosphoinositides in murine epidermal keratinocytes. J Cell
Physiol 132:131–136
Kim H-J, Abdelkader N, Katz M, McLane JA 1992 1,25 Dihydroxyvitamin D3 enhances antiproliferative effect and transcription of TGF-b1 on human keratinocytes in culture. J Cell Physiol
151:579 –587
Kobayashi T, Hashimoto K, Yoskikawa K 1993 Growth inhibition
of human keratinocytes by 1,25-dihydroxyvitamin D3 is linked to
dephosphorylation of retinoblastoma gene product. Biochem Biophys Res Commun 196:487– 493
Yen A, Chandler S, Forbes ME, Fung YK, Tang A, Pearson R 1992
Coupled down regulation of the RB retinoblastoma and c-myc
genes antecede cell differentiation: possible role of RB as a status
quo gene. Eur J Cell Biol 57:210 –221
Manfredini R, Grande A, Tagliafico E, Barbieri D, Zucchini P, Citro
G, Zupi G, Franceschi C, Torelli U, Ferrari S 1993 Inhibition of c-fos
expression by an anti-sense oligomer causes apoptosis of HL60 cells
induced to granulocytic differentiation. J Exp Med 178:381–389
Studzinski GP, Bhandal AK, Brelvi ZS 1985 Cell cycle sensitivity
of HL-60 cells to the differentiation-inducing effects of 1a,25dihydroxyvitamin D3. Cancer Res 45:3898 –3905
Wang QM, Jones JB, Studzinski GP 1996 Cyclin-dependent kinase
inhibitor p27 as a mediator of the G1-S phase block induced by
1,25-dihydroxyvitamin D3 in HL60 cells. Cancer Res 56:264 –267
Jiang H, Lin J, Su Z-Z, Collart FR, Huberman E, Fisher PB 1994
Induction of differentiation in human promyleocytic HL-60 leukemia cells activates, p21, WAF1/CIP1, expression in the absence
of p53. Oncogene 9:3397–3406
Zhang W, Grasso L, McClain GD, Gambel AM, Cha Y, Travali S,
Deisseroth AB, Mercer WE 1995 p53-independent induction of
WAF/CIP1 in human leukemia cells is correlated with growth
arrest accompanying monocyte/macrophage differentiation. Cancer Res 55:668 – 674
Wang QM, Luo X, Studzinski GP 1997 Cyclin-dependent kinase
6 is the principal target of p27/Kip1 regulation of the G1-phase
traverse in 1,25-dihydroxyvitamin D3-treated HL60 cells. Cancer
Res 57:2851–2855
Hengst L, Reed SI 1996 Translational control of p27Kip1 accumulation during the cell cycle. Science 271:1861–1864
Liu M, Lee M-H, Cohen M, Bommakanti M, Freedman LP 1996
Transcriptional activation of the Cdk inhibitor p21 by vitamin D3
leads to the induced differentiation of the myelomonocytic cell line
U937. Genes Dev 10:142–153
Wilhide C, Van Dang C, Dipersio J, Kenedy AA, Bray PF 1995
Overexpression of cyclin D1 in the Dami megakaryocytic cell line
causes growth arrest. Blood 86:294 –304
Bruchovsky N, Lesser B, Van Dorne E, Craven S 1975 Hormonal
effects on cell proliferation in rat prostate. Vitam Horm 33:61–102
Kyprianou N, Isaaca JT 1988 Activation of programmed cell death
in the rat ventral prostate after castration. Endocrinology 122:
552–562
Katz AE, Benson MC, Wise GJ, Olsson CA, Bandyk MG, Sawc-
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
532
359.
360.
361.
362.
363.
364.
365.
366.
367.
368.
369.
370.
371.
372.
373.
374.
375.
376.
377.
378.
379.
PESTELL ET AL.
zuki IS, Tomashefsky P, Buttyan R 1989 Gene activity during the
early phase of androgen-stimulated rat prostate regrowth. Cancer
Res 49:5889 –5894
Furuya Y, Lin XS, Walsh JC, Nelson WG, Isaacs JT 1995 Androgen
ablation-induced programmed death of prostatic glandular cells
does not involve recruitment into a defective cell cycle or p53
induction. Endocrinology 136:1898 –906
Chen Y, Robles AI, Martinez LA, Gimenez-Conti IB, Conti CJ
1996 Expression of the G1 cyclins, cyclin-dependent kinases, and
cyclin-dependent kinase inhibitors in androgen-induced prostate
proliferation in castrated rats. Cell Growth Differ 7:1571–1578
Lu S, Tsai SY, Tsai M-J 1997 Regulation of androgen-dependent
prostatic cancer cell growth: androgen regulation of Cdk2, Cdk4 and
CKIp16 genes. Cancer Res 57:4511– 4516
Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM,
Evans RM 1995 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand
for the adipocyte determination factor PPARg. Cell 83:803– 812
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkinson WO, Willson TM, Kliewer SA 1995 An antidiabetic thiazolidinedione is a
high affinity ligand for peroxisome proliferator-activated receptor
gamma (PPAR gamma). J Biol Chem 270:12953–12956
Kliewer SA, Lenhard JM, Wilson TM, Patel I, Morris DC, Lehmann JM 1995 A prostaglandin J2 metabolite binds peroxisome
proliferator-activated receptor gamma and promotes adipocyte
differentiation. Cell 83:813– 819
Mandrup S, Lane MD 1997 Regulating adipogenesis. Minireview.
J Biol Chem 272:5367–5370
Tontonoz P, Kim JB, Graves RA, Spiegelman BM 1993 A novel
helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol 13:4753– 4759
Altiok S, Xu M, Spiegelman BM 1998 PPARg induces cell cycle
withdrawal:inhibition of E2F/DP DNA-binding activity via downregulation of PP2A. Genes Dev 11:1987–1998
Boynton AL, Whitfield JF 1983 The role of cyclic AMP in cell
proliferation: a critical assessment of the evidence. Adv Cyclic
Nucleotide Res 15:193–294
Dumont JE, Jaunieux J-C, Roger PP 1989 The cyclic AMP-mediated
stimulation of cell proliferation. Trends Biochem Sci 14:67–71
Grieco D, Porcellini A, Avvedimento EV, Gottesman ME 1996
Requirement for cAMP-PKA pathway activation by M phase promoting factor in the transition from mitosis to interphase. Science
271:1718 –1723
Florio T, Scorizello A, Fattore M, D’Alto V, Salzano S, Rossi G,
Berlingieri MT, Fusco A, Schettini G 1996 Somatostatin inhibits
PC C13 thyroid cell proliferation through the modulation of phosphotyrosine activity. Impairment of the somatostatinergic effects
by stable expression of E1A viral oncoprotein. J Biol Chem 271:
6129 – 6136
Struthers RS, Vale WW, Arias C, Sawchenko PE, Montminy MR
1991 Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature 350:622–624
Vairo G, Argyriou S, Bordun A-M, Whitty G, Hamilton JA 1990
Inhibition of signalling pathways for macrophage proliferation by
cyclic AMP. J Biol Chem 265:2692–2701
Vadiveloo PK, Vairo G, Royston Novak AK, Whitty G, Filonzi EL,
Cragoe EJJ, Hamilton JA 1996 Differential regulation of cell-cycle
machinary by various antiproliferative agents is linked to macrophage arrest at distinct G1 checkpoints. Oncogene 13:599 – 608
Cocks B, Vairo G, Bodrug SE, Hamilton J 1992 Suppression of
growth factor-induced CYL1 cyclin gene expression by antiproliferative agents. J Biol Chem 267:2307–2310
Kato J-y, Matsuoka M, Polyak K, Massague Sherr CJ 1994 Cyclic
AMP-induced G1 phase arrest mediated by an inhibitor of cyclindependent kinase 4 activation. Cell 79:487– 496
L’Allemain G, Lavoie JN, Rivard N, Baldin V, Pouyssegur J 1997
Cyclin D1 expression is a major target of the cAMP-induced inhibition of cell cycle entry in fibroblasts. Oncogene 14:1981–1990
Desdouets C, Matesic G, Molina CA, Poulkes NS, Sassone-Corsi
P, Brechot C, Sobczak-Thepot J 1995 Cell cycle regulation of cyclin
A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol Cell Biol 15:3301–3309
Yoshizumi M, Hsieh CM, Zhou F, Tsai JC, Patterson C, Perrella
MA, Lee ME 1995 The ATF site mediates downregulation of the
380.
381.
382.
383.
384.
385.
386.
387.
388.
389.
390.
391.
392.
393.
394.
395.
396.
397.
398.
399.
400.
Vol. 20, No. 4
cyclin A gene during contact inhibition in vascular endothelial
cells. Mol Cell Biol 15:3266 –3272
Barlat I, Henglein B, Plet A, Lamb N, Fernandez A, McKenzie F,
Pouyssegur J, Vie A, Blanchard JM 1995 TGF-b1 and cAMP attenuate cyclin A gene transcription via a cAMP responsive element
through independent pathways. Oncogene 11:1309 –1318
Lamas M, Molina C, Foulkes NS, Jansen E, Sassone-Corsi P 1997
Ectopic ICER expression in pituitary corticotroph AtT20 cells: effect
on morphology, cell cycle and hormonal regulation. Mol Endocrinol 11:1425–1434
Yamashita YM, Nakaseko Y, Samejima I, Kumada K, Yamada H,
Michaelson D, Yanagida M 1996 20S cyclosome complex formation and proteolytic activity inhibited by the cAMP/PKA pathway.
Nature 384:276 –279
Hunter T, Karin M 1992 The regulation of transcription by phosphorylation. Cell 70:375–387
Yuspa S, Hennings H, Kulesz-Martin M, Lichti U 1982 The study
of tumor promotion in a cell culture model for mouse skin–a tissue
that exhibits multistage carcinogenesis in vivo. In: Hecker E,
Fusenig NE, Kunz W, Marks F, Thielman HW (eds) Cocarcinogenesis and Biological Effects of Tumor Promoters. Raven Press,
New York, pp 217–229
Huang T-S, Duyster J, Wang JYJ 1995 Biological response to phorbol ester determined by alternative G1 pathways. Proc Natl Acad
Sci USA 92:4793– 4797
Zeng Y-X, Somasundaram K, El-Deiry WS 1997 AP2 inhibits cancer cell growth and activates p21WAF/CIP1 expression. Nat Genet
15:78 – 82
Williams T, Admon A, Luscher B, Tjian R 1988 Cloning and
expression of AP-2, a cell-type-specific transcription factor that
activates inducible enhancer elements. Genes Dev 2:1557–1569
Hiroaki Kiyokawa H, Kineman RD, Manova-Todorova KO,
Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC,
Frohman LA, Koff A 1996 Enhanced growth of mice lacking the
cyclin-dependent kinase inhibitor function of p27. Cell 85:721–732
Geradts J, Wilson PA 1996 High frequency of aberrant p16 INK4A
expression in human breast cancer. Am J Pathol 149:15–20
Hosokawa Y, Tu T, Tahara H, Smith AP, Arnold A 1995 Absence
of cyclin D1/PRAD1 point mutations in human breast cancers and
parathyroid adenomas and identification of a new cyclin D1 gene
polymorphism. Cancer Lett 93:165–170
Hsi ED, Zukerberg LR, Yang WI, Arnold A 1996 Cyclin D1/
PRAD1 expression in parathyroid adenomas: an immunohistochemical study. J Clin Endocrinol Metab 81:1736 –1739
Lloyd RV, Jin L, Qian X, Kulig E 1997 Aberrant p27 kip1 expression in endocrine and other tumors. Am J Pathol 150:401– 407
Tahara H, Smith AP, Gaz RD, Arnold A 1996 Loss of chromosome
arm 9p DNA and analysis of the p16 and p15 cyclin-dependent
kinase inhibitor genes in human parathyroid adenomas. J Clin
Endocrinol Metab 81:3663–3667
Cryns VL, Thor A, Xu HJ, Hu SX, Wierman ME, Vickery ALJ,
Benedict WF, Arnold A 1994 Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med 330:757–761
Dotzenrath C, Teh BT, Farnebo F, Cupisti K, Svensson A, Toell
A, Goretzki P, Larsson C 1996 Allelic loss of the retinoblastoma
tumor suppressor gene: a marker for aggressive parathyroid tumors? J Clin Endocrinol Metab 8:3194 –3196
Ledent C, Dumont J, Vassart G, Parmentier M 1991 Thyroid adenocarcinomas secondary to tissue-specific expression of simian
virus-40 large T-antigen in transgenic mice. Endocrinology 129:
1391– 401
Ledent C, Marcotte A, Dumont JE, Vassart G, Parmentier M 1995
Differentiated carcinomas develop as a consequence of the thyroid
specific expression of a thyroglobulin-human papillomavirus type
16 E7 transgene. Oncogene 10:1789 –1797
Ito Y, Kobayashi T, Takeda T, Komoike Y, Wakasugi E, Tamaki
Y, Tsujimoto M, Matsuura N, Monden M 1996 Expression of p21
(WAF1/CIP1) protein in clinical thyroid tissues. Br J Cancer 74:
1269 –1274
Shi Y, Zou M, Farid NR, al-Sedairy ST 1996 Evidence of gene
deletion of p21 (WAF1/CIP1), a cyclin-dependent protein kinase
inhibitor, in thyroid carcinomas. Br J Cancer 74:1336 –1341
Goretzki PE, Gorelov V, Dotzenrath C, Witte J, Roeher HD 1996
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
August, 1999
401.
402.
403.
404.
405.
406.
407.
408.
409.
410.
411.
412.
413.
414.
415.
416.
417.
418.
419.
420.
CYCLINS AND CYCLIN INHIBITORS IN ENDOCRINE SIGNALING
A frequent mutation/polymorphism in tumor suppressor gene
INK4b (MTS-2) in papillary and medullary thyroid cancer. Surgery
120:1081–1088
Tanaka C, Yoshimoto K, Yang P, Kimura T, Yamada S, Moritani
M, Sano T, Itakura M 1997 Infrequent mutations of p27Kip1 gene
and trisomy 12 in a subset of human pituitary adenomas. J Clin
Endocrinol Metab 82:3141–3147
Jin L, Qian X, Kulig E, Sanno N, Scheithauer BW, Kovacs K, Young
WFJ, Lloyd RV 1997 Transforming growth factor-b, transforming
growth factor-b receptor II, and p27Kip1 expression in nontumorous
and neoplastic human pituitaries. Am J Pathol 151:509 –519
Jacks T, Fazel A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA 1992 Effects of an Rb mutation in the mouse. Nature
359:295–300
Hu N, Gutsmann A, Herbert DC, Bradley A, Lee W-H, Lee EY 1994
Heterozygous Rb-1 delta 20/1 mice are predisposed to tumors of
the pituitary gland with a nearly complete penetrance. Oncogene
9:1021–1027
Yamasaki L, Bronson R, Williams B, Dyson NJ, Harlow E, Jacks
T 1998 Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(6) mice. Nat Genet 18:360 –364
Cryns VL, Alexander JM, Klibanski A, Arnold A 1993 The retinoblastoma gene in human pituitary tumors. J Clin Endocrinol
Metab 77:644 – 646
Zhu J, Leon SP, Beggs AH, Busque L, Gilliland DG, Black PM
1994 Human pituitary adenomas show no loss of heterozygosity at
the retinoblastoma gene locus. J Clin Endocrinol Metab 78:922–927
Pei L, Melmed S, Scheithauer B, Kovacs K, Benedict WF, Prager
D 1995 Frequent loss of heterozygosity at the retinoblastoma susceptibility gene (RB) locus in aggressive pituitary tumors: evidence
for a chromosome 13 tumor suppressor gene other than RB. Cancer
Res 55:1613–1616
Levy A, Hall L, Yeundall WA, Lightman SL 1994 p53 Gene mutations in pituitary adenomas: rare events. Clin Endocrinol (Oxf)
41:809 – 814
Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhowev R,
Chen-Kinag S, Su L, Xiong Y 1998 Cdk inhibitors p18INK4c and
p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 12:2899 –2911
Asa SL, Ezzat S 1998 The cytogenesis and pathogenesis of pituitary
adenomas. Endocr Rev 19:798 – 827
Lee MP, DeBaun M, Randhawa G, Reichard BA, Elledge SJ,
Feinberg AP 1997 Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am J Hum Genet 61:304 –309
Liu J, Kahri AI, Heikkila P, Voutilainen R 1997 Ribonucleic acid
expression of the clustered imprinted genes, p57KIP2, insulin-like
growth factor II, and H19, in adrenal tumors and cultured adrenal
cells. J Clin Endocrinol Metab 82:1766 –1771
Shigemasa K, Hu C, West CM, Clarke J, Parham GP, Parmley TH,
Korourian S, Baker VV, O’Brien TJ 1997 p16 Overexpression: a
potential early indicator of transformation in ovarian carcinoma. J
Soc Gynecol Invest 4:95–102
Hatta Y, Hirama T, Takeuchi S, Lee E, Pham E, Miller CW,
Strohmeyer T, Wilczynski SP, Melmed S, Koeffler HP 1995 Alterations of the p16 (MTS1) gene in testicular, ovarian, and endometrial malignancies. J Urol 154:1954 –1957
Lukas J, Groshen S, Saffari B, Niu N, Reles A, Wen W, Felix J,
Jones LA, Hall FL, Press MF 1997 WAF1/Cip1 gene polymorphism
and expression in carcinomas of the breast, ovary, and endometrium. Am J Pathol 150:167–175
Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ
1996 Tumor induction and tissue atrophy in mice lacking E2F-1.
Cell 85:537–548
Field SJ, Tsai F-Y, Kuo F, Zubiaga AM, Kaelin WGJ, Livingston
DM, Orkin SH, Greenberg ME 1996 E2F-1 functions in mice to
promote apoptosis and suppress proliferation. Cell 85:549 –561
Houldsworth J, Reuter V, Bosl GJ, Chaganti RS 1997 Aberrant
expression of cyclin D2 is an early event in human male germ cell
tumorigenesis. Cell Growth Differ 8:293–299
Tsujimoto Y, Yunis J, Onorato-Showe L, Erikson J, Nowell PC,
Croce CM 1984 Molecular cloning of the chromosomal breakpoint
of B-cell lymphomas and leukemias with the t(11;14) chromosome
translocation. Science 224:1403–1406
533
421. Komatsu H, Iida S, Yamamoto K, Mikuni C, Nitta M, Takahashi
T, Ueda R, Seto M 1994 A variant chromosome translocation at
11q13 identifying PRAD1/cyclin D1 as the BCL-1 gene. Blood
84:1226 –1231
422. Williams ME, Swerdlow SH, Rosenberg CL, Arnold A 1993 Chromosome 11 translocation breakpoints at the PRAD1/cyclin D1 gene
locus in centrocytic lymphoma. Leukemia 7:241–245
423. Lammie GA, Peters G 1991 Chromosome 11q13 abnormalities in
human cancer. Cancer Cells 3:413– 420
424. Akervall JA, Michalides RJ, Mineta H, Balm A, Borg A, Dictor
MR, Jin Y, Loftus B, Mertens F, Wennerberg JP 1997 Amplification
of cyclin D1 in squamous cell carcinoma of the head and neck and
the prognostic value of chromosomal abnormalities and cyclin D1
overexpression. Cancer 79:380 –389
425. Gansauge S, Gansauge F, Ramadani M, Stobbe H, Rau B, Harada
N, Beger HG 1997 Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res 57:
1634 –1637
426. Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth
RF, Wittenbel KD, Simpson JF, Page DL, Steeg PS 1995 Overexpression of cyclin D mRNA distinguishes invasive and in situ breast
carcinomas from non-malignant lesions. Nature Med 1:1257–1259
427. van Diest P, Michalides RJ, Jannink L, van der Valk P, Peterse HL,
de Jong JS, Meijer CJ, Baak JP 1997 Cyclin D1 expression in
invasive breast cancer. Correlations and prognostic value. Am J
Pathol 150:705–711
428. Seshadri R, Lee CS, Hui R, McCaul K, Horsfall DJ, Sutherland RL
1996 Cyclin D1 amplification is not associated with reduced overall
survival in primary breast cancer but may predict early relapse in
patients with features of good prognosis. Clin Cancer Res 2:1177–1184
429. Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA 1994
Function of human cyclin gene as an oncogene. Proc Natl Acad Sci
USA 91:709 –713
430. Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt
EV 1994 Mammary hyperplasia and carcinoma in MMTV-cyclin D1
transgenic mice. Nature 369:669 – 671
431. Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW,
Adams JM 1994 Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene.
EMBO J 13:2124 –2130
432. Lovec H, Grzeschiczek A, Kowalski MB, Moroy T 1994 Cyclin
D1/bcl-1 cooperates with myc genes in the generation of B-cell
lymphoma in transgenic mice. EMBO J 13:3487–3495
433. Nakagawa H, Wang TC, Zukerberg L, Odze R, Togawa K, May
GHW, Wilson J, Rustgi AK 1997 The targeting of the cyclin D1
oncogene by an Epstein-Barr virus promoter in transgenic mice
causes dysplasia in the tongue, esophagus and forestomach. Oncogene 14:1185–1190
434. Robles AI, Larcher F, Whalin RB, Murillas R, Richie E, GimenezConti IB, Jorcano JL, Conti CJ 1996 Expression of cyclin D1 in
epithelial tissues of transgenic mice results in epidermal hyperproliferation and severe thymic hyperplasia. Proc Natl Acad Sci
USA 93:7634 –7638
435. Liu J-J, Chao J-R, Jiang M-C, Ng S-Y, Yen J J- Y, Yang-Yen H-F 1995
Ras transformation results in an elevated level of cyclin D1 and
acceleration of G1 progression in NIH3T3 cells. Mol Cell Biol 15:
3654 –3663
436. Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L,
Sicinski P, Tennant RW, Weinbergh RA, Yuspa SH, Conti CJ 1998
Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev 12:2469 –2474
437. Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim N-H, Delohery T, Klein MG, Holt PR, Weinstein IB 1997 Antisense to
cyclin D1 inhibits the growth and tumorigenicity of human colon
cancer cells. Cancer Res 57:1569 –1574
438. Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L,
Pestell RG, Der CJ 1997 Rac regulation of transformation, gene
expression and actin organisation by multiple, PAK-independent
pathways. Mol Cell Biol 17:1324 –1335
439. Watanabe G, Howe A, Lee RJ, Albanese C, Shu I-W, Karnezis A,
Zon L, Kyriakis J, Rundell K, Pestell RG 1996 Induction of cyclin
D1 by simian virus 40 small tumor antigen. Proc Natl Acad Sci USA
93:12861–12866
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.
534
PESTELL ET AL.
440. Westwick JK, Lee RJ, Lambert QT, Pestell RG, Kay R, Der CJ,
Whitehead IP 1998 Transforming potential of Dbl family proteins
correlates with transcription from the cyclin D1 promoter but not
with activation of Jun NH2-terminal kinase, p38/Mpk2, serum
response factor, or c-Jun. J Biol Chem 273:16739 –16744
441. Rosenwald IB, Kaspar R, Rousseau D, Gehrke L, Leboulch P,
Chen JJ, Schmidt EV, Sonenberg N, London IM 1995 Eukaryotic
translation initiation factor 4E regulates expression of cyclin D1 at
transcriptional and post-transcriptional levels. J Biol Chem 270:
21176 –21180
442. Rosenwald IB, Lazaris-Karatus A, Sonenberg N, Schmidt EV 1993
Elevated levels of cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Mol Cell Biol 13:7358–7363
443. Sawa H, Ohshima TA, Ukita H, Murakami H, Chiba Y, Kamada
H, Hara M, Saito I 1998 Alternatively spliced forms of cyclin D1
modulate entry into the cell cycle in an inverse manner. Oncogene
16:1701–1712
444. Zhou P, Jiang W, Weghorst CM, Weinstein IB 1996 Overexpression of cyclin D1 enhances gene amplification. Cancer Res 56:36 –39
445. Meijer L 1996 Chemical inhibitors of cyclin-dependent kinases.
Trends Cell Biol 6:393–397
446. Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R 1996
Genetic selection of peptide aptamers that recognize and inhibit
cyclin-dependent kinase 2. Nature 380:548 –550
447. Henglein B, Chenivesse X, Wang J, Eick D, Brechot C 1994 Structure and cell cycle-regulated transcription of the human cyclin A
gene. Proc Natl Acad Sci USA 91:5490 –5494
448. Geng Y, Eaton EN, Picon M, Roberts JM, Lundberg AS, Gifford
A, Sardet C, Weinberg RA 1996 Regulation of cyclin E transcription
by E2Fs and retinoblastoma protein. Oncogene 12:1173–1180
449. Walker SS, Shen WC, Reese JC, Apone LM, Green MR 1997 Yeast
TAF(II)145 required for transcription of G1/S cyclin genes and
regulated by the cellular growth state. Cell 90:607– 614
450. Sekiguchi T, Nohiro Y, Nakamura Y, Hasamato N, Nishimoto T
1991 The human CCG1 gene, essential for progression of the G1
phase, encodes a 210-kilodalton nuclear DNA-binding protein. Mol
Cell Biol 11:3317–3325
451. Wang EH, Zou S, Tjian R 1997 TAFII250-dependent transcription
of cyclin A is directed by ATF activator proteins. Genes Dev 11:
2658 –2669
452. Ramakrishnan M, Musa NL, Li J, Liu PT, Pestell RG, Hershenson
MB 1998 Catalytic activation of extracellular signal-regulated kinases regulates cyclin D1 expression in primary tracheal myocytes.
Am J Respir Cell Mol Biol 18:736 –740
453. Lee RJ, Watanabe G, Albanese C, Reutens AT, Haines GKI, Siegel
PM, Muller WJ, Pestell RG 1998 Regulation of cyclin D1 by the
Neu, c-erbB2, proto-oncogene. Proceedings of the 8th Annual Meeting of the American Association for Cancer Research, New Orleans,
LA, 1998 (Abstract 1730)
454. Herber B, Truss M, Beato M, Muller R 1994 Inducible regulatory
elements in the human Cyclin D1 promoter. Oncogene 4:1295–1304
455. Lee RJ, Albanese C, Stenger RJ, Watanabe G, Inghirami G,
Haines III GK, Webster M, Muller WJ, Brugge JS, Davis R, Pestell
RG 1999 pp60v-src Induction of cyclin D1 requires collaborative
interactions between the ERK, p38 and Jun kinase pathways: A role
for CREB and ATF-2 in pp60v-src signaling in breast cancer cells.
J Biol Chem 274:7341–7350
456. Beier F, Lee RJ, Taylor A, Pestell RG, LuValle P 1999 Identification
of the cyclin D1 gene as a target of ATF-2 in chondrocytes. Proc Natl
Acad Sci USA 99:1433–1438
457. Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, Pestell
RG, Greenberg ME 1998 Fos family members induce cell cycle
entry by activating cyclin D1. Mol Cell Biol 18:5609 –5619
458. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M,
Pestell RG, Ben-Ze’ev A 1999 The cyclin D-1 gene is a target of the
b-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 96:5522–5527
459. Tetsu O, McCormick F 1999 b-Catenin regulates expression of
cyclin D1 in colon carcinoma cells. Nature 398:422– 424
460. Matsumura I, Kitamura T, Wakao H, Tanaka H, Hashimoto K,
Albanese C, Downward J, Pestell RG, Kanakura Y 1999 Transcriptional regulation of cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO
J 18:101–111
Vol. 20, No. 4
461. Watanabe G, Albanese C, Lee RJ, Reutens A, Vairo G, Henglein
B, Pestell RG 1998 Inhibition of cyclin D-kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity
through E2F and Sp1. Mol Cell Biol 18:3212–3222
462. Steiner P, Philipp A, Godden-Kent D, Pagano M, Mittnacht S,
Bartek J, Eilers M 1995 Identification of a myc-dependent step
during the formation of active G1 cyclin-Cdk complexes. EMBO J
14:4814 – 4826
463. Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ 1994 Myc
induces cyclin D1 expression in the absence of de novo protein
synthesis and links mitogen-stimulated signal transduction to the
cell cycle. Oncogene 9:3635–3645
464. Muller H, Lukas J, Schneider A, Warthoe P, Bartek J, Eilers M,
Strauss M 1994 Cyclin D1 expression is regulated by the retinoblastoma protein. Proc Natl Acad Sci USA 91:2945–2949
465. Jansen-Durr P, Meichle A, Steiner P, Pagano M, Finke K, Botz J,
Wessbecher J, Draetta G, Eilers M 1993 Differential modulation of
cyclin gene expression by MYC. Proc Natl Acad Sci USA 90:3685–3689
466. Chen X, Bargonetti J, Prives C 1995 p53, Through p21 (WAF1/
CIP1), induces cyclin D1 synthesis. Cancer Res 55:4257– 4263
467. Atadja P, Wong H, Veillete C, Riabowal K 1995 Overexpression
of cyclin D1 blocks proliferation of normal diploid fibroblasts. Exp
Cell Res 217:205–216
468. Brooks AR, Shiffman D, Chan CS, Brooks EE, Milner PG 1996
Functional analysis of the human cyclin D2 and cyclin D3 promoters. J Biol Chem 271:9090 –9099
469. Yang M, Hosokawa Y, Kaneko S, Tanaka M, Nakashima K 1996
Structure and characterization of rat cyclin D3 promoter. Gene
181:153–159
470. Wang Z, Sicinski P, Weinberg RA, Zhang Y, Ravid K 1996 Characterization of the mouse cyclin D3 gene: exon/intron organization
and promoter activity. Genomics 35:156 –163
471. Yoshizumi M, Wang H, Hsieh CM, Sibinga NE, Perrella MA, Lee
ME 1997 Down-regulation of the cyclin A promoter by transforming growth factor-b1 is associated with a reduction in phosphorylated activating transcription factor-1 and cyclic AMP-responsive
element-binding protein. J Biol Chem 272:22259 –22264
472. Brehm A, Miska E, McCance D, Reid J, Bannister A, Kouzarides
T 1998 Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597– 601
473. Alland L, Muhle R, Hou HJ, Potes J, Chin L, Schreiber-Agus N,
DePinho RA 1997 Role for N-CoR and histone deacetylase in
Sin3-mediated transcriptional repression. Nature 387:49 –55
474. Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth
K, Roberts JM, Ross R 1998 Cleavage of p21Cip1/Waf1 and p27Kip1
mediates apoptosis in endothelial cells through activation of Cdk2:
role of a caspase cascade. Mol Cell 1:553–563
475. Matsuoka M, Kato J, Fisher RP, Morgan DO, Sherr CJ 1994 Activation of cyclin-dependent kinase-4 (Cdk4) by mouse MO15 associated kinase. Mol Cell Biol 14:7265–7275
476. Byeon I-JL, Li J, Ericson K, Selby TL, Tevelev A, Kim H-J,
O’Maille P, Tsai M-D 1998 Tumor suppressor p16Ink4a: determination of solution structure and analyses of its interaction with
cyclin-dependent kinase 4. Mol Cell 1:421– 431
477. Lundberg AS, Weinberg RA 1998 Functional inactivation of the
retinoblastoma protein requires sequential modification by at least
two distinct cyclin-Cdk complexes. Mol Cell Biol 18:753–761
478. Schulze A, Zerfass K, Spitkovsky D, Middendorp S, Berges J,
Helin K, Jansen-Durr P, Henglein B 1995 Cell cycle regulation of
cyclin A gene promoter is mediated by a variant E2F site. Proc Natl
Acad Sci USA 92:11264 –11268
479. Zwicker J, Liu N, Engeland K, Lucibello FC, Muller R 1996 Cell cycle
regulation of E2F site occupation in vivo. Science 271:1595–1597
480. Huet X, Rech J, Plet A, Vie A, Blanchard JM 1996 Cyclin A expression is under negative transcriptional control during the cell
cycle. Mol Cell Biol 16:3789 –3798
481. Bromberg J, Devgan G, Zhao Y, Albanese C, Pestell RG, Darnell
J, Stat3 as an oncogene. Cell, in press
482. Guttridge D, Albanese C, Reuther JY, Pestell RG, Baldwin Jr AS,
NF-KB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19:5785–5799
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 04 March 2016. at 15:40 For personal use only. No other uses without permission. . All rights reserved.