Chapter 8 Problems Page 1 of 6 11/1/2007 8.1 The decarboxylation
advertisement

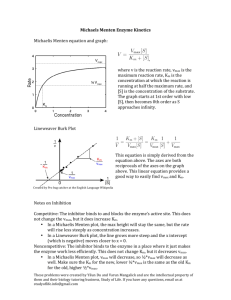
Chapter 8 Problems Page 1 of 6 11/1/2007 8.1 The decarboxylation of a beta-keto acid catalyzed by an enzyme can be measured by the rate of formation of CO2. From the initial rates in the table, determine the MichaelisMenten constant for the enzyme and the maximal velocity by a graphical method. First, the problem should really say “determine KM and Vmax for this enzyme-substrate combination”. The values for these constants depend on what enzyme and what substrate are being considered. I chose to transform the data for an Eadie-Hofstee plot (v0/[S] vs. v0). For this plot, the slope is -1/KM and the vertical axis intercept is Vmax/KM. The plot is below. From Excel, I get the least-squares values for the slope (-2.26 M-1) and intercept (1.57 μmol/2min·M). (I left the values in the units that were in the table.) From the slope, we find that KM = 1/(-2.26 M-1) = 0.442 M. From the intercept, Vmax = KM·intercept = (0.442 M)·(1.57 μmol/2min·M) = 0.695 μmol/2min = 0.347 μmol/min. 1.8 Eadie-Hofstee plot 1.6 1.4 1.2 v0/[S] 1 0.8 0.6 0.4 0.2 0 0 0.1 0.2 0.3 0.4 v0 0.5 0.6 0.7 Chapter 8 Problems Page 2 of 6 11/1/2007 8.5. An enzymatic reaction has a simple Michaelis-Menten mechanism: k1 k2 E + S ↔ ES → E + P k-1 k1 and k-1 are very fast, k2 = 100 1/sec and KM = 10-4 M at 280 K while k2 = 200 1/sec and KM = 1.5 × 10-4 M at 300 K. (a) For [S] = 0.1 M and [E]0 = 10-5 M, what is v0 at 280 K? (b) Calculate the activation energy for k2. (c) What is Keq at 280 K for E binding to S? (d) What is ΔH0 for the formation of ES from E and S? a) We are given KM, and need to calculate Vmax = k2·[E]0 = (100 1/sec)( 10-5 M) = 10-3 M/s. Micaelis-Menten gives us v0 = Vmax [ S ] (10 −3 M / s )(0.1M ) = = 10 − 3 M / s −4 K M + [S ] 10 M + 0.1M b) From the Arrhenius relationship k = Ae − E A / RT we can divide k2 at T = 280 K by k2’ at 300 K (I denote the different Ts by a prime on the second k) and then take the logarithm of the ratio: k E ⎛1 1⎞ EA 1 ⎞ 100 s −1 ⎛ 1 −5 ln 2 = ln = −.693 = − A ⎜ − ⎟ = − − ⎟ = −(2.86 × 10 mol / J ) E A ⎜ ' −1 R ⎝T T'⎠ 8.314 J / mol • K ⎝ 280 K 300 K ⎠ k2 200 s so from this we have EA = (0.693)/(2.86 × 10-5 mol/J) = 24.2 kJ/mol. c) Keq= k1/k-1 by definition. KM = (k-1 + k2)/k1 ≅ k-1/k1 since k-1 >> k2 from the problem statement that the first reaction is very fast. Thus, Keq = 1/KM = 1/(10-4 M) = 104 M-1. d) This problem is asking about how enthalpy of a reaction depends on the change of the equilibrium constant with temperature. This is the van’t Hoff relationship: ΔH 0 = − R ln(10 −4 ) − ln(1.5 ×10 −4 ) (−9.21) − (−8.80) Δ(ln K ) = −R = −(8.314 J / K • mol ) = −114.2kJ / mol −3 −3 1 1 1 ( ) Δ( ) − ( 3 . 57 10 3 . 33 10 ) × − × 280 K 300 K T Chapter 8 Problems Page 3 of 6 11/1/2007 8.10. Penicillinase inactivates penicillin. One form with MW = 30,000 has a single active site, kcat = 2000 s-1, and KM = 5 × 10-5 M. In response to treatment with 5 μmol of penicillin, a 1 mL suspension of bacteria release 0.04 μg of enzyme. (a) Assuming rapid mixing and equilibration, what fraction of E will complex with penicillin early in the reaction? (b) How long will it take for half of the initial amount of penicillin to be inactivated? (c) What concentration of penicillin would cause the enzyme to react at half maximal velocity? (d) A second suspension of bacteria releases 0.06 μg/mL of enzyme. Will this affect the answers to (b) and (c) and, if so, by how much? (e) Modified penicillin acts as a competitive inhibitor. If the affinity of E for penicillin and modified penicillin is the same, what concentration of the inhibitor will reduce the rate of loss of penicillin fivefold at low [penicillin]? [ ES ] [ ES ] 1 = = where [ ES ] + [ E ] [ ES ] + K M [ ES ] 1 + K M [S ] [S ] -5 the second step comes from KM = [E][S]/[ES]. Plugging in KM = 5 × 10 M and [S] = 5 μmol/1 mL = 5 × 10-3 M, we get f = 0.999. a) The fraction of E complexed to S is f = b) We can see that, from the M-M equation, when [S] = 2.5 × 10-3 M, v0 is still 0.98Vmax, so the system will remain in the approximate zero-order domain. The velocity during this time will be equal to Vmax to a good approximation. Vmax = kcat[E]0 = (2000 s-1)(0.04 μg/(30,000 g/mol)/(0.001 L) = 2.67 μM/s. The change in the concentration of S is -2.5 × 10-3 M, so Δ[S]/Δt = -2.67 μM/s, or Δt = ( -2.5 × 10-3 M)/(-2.67 μM/s) = 937 sec = 15.6 min. c) v0 = Vmax/2 when [S] = KM, so [penicillin] = 5 × 10-5 M. d) Yes, this will affect part b) because [E]0 increases by 50%, so Vmax increases by 50% and all velocities will thus increase by this factor. That means that Δt will decrease by a factor of 1.5, or Δt = 625 sec. Part c) won’t change since the affinity doesn’t depend on how much E is around. e) If the inhibitor has the same affinity as penicillin, then KI = KM = 5 × 10-5 M. For a competitive inhibitor we have Vmax [ S ] . So when v0(uninhibited) = 5v0(inhibited), we have v0 = ⎛ [I ] ⎞ ⎟⎟ + [ S ] K M ⎜⎜1 + K I ⎠ ⎝ Vmax [ S ] 5Vmax [ S ] 1 5 . That is, = or = K M + [S ] ⎛ ⎛ ⎛ 0 ⎞ [I ] ⎞ [I ] ⎞ ⎟⎟ + [ S ] ⎟⎟ + [ S ] K M ⎜⎜1 + ⎟⎟ + [ S ] K M ⎜⎜1 + K M ⎜⎜1 + ⎝ KI ⎠ ⎝ KM ⎠ ⎝ KI ⎠ K ⎛ [ I ] ⎞ [S ] ⎟+ so [ I ] = 4(K M + [ S ]) , thus it depend on how much S K M + [ S ] = M ⎜⎜1 + K M ⎟⎠ 5 5 ⎝ is present. If [S] << KM, then [I] = 4KM = 2 × 10-4 M. Chapter 8 Problems Page 4 of 6 11/1/2007 8.16. RNA polymerase binds rapidly to DNA, then the complex rearranges slowly. This can be diagrammed as k1 A+B ↔ C k2 k-1 C ↔ D k -2 fast slow At the start of the reaction 1 nM A and 1 nM B are present. At final equilibrium the concentrations are [A] = [B] = 0.6 nM, [C] = 0.01 nM and [D] = 0.39 nM. (a) Calculate K1 and K2 from the equilibrium concentrations. (b) Write the differential equations for d[C]/dt and d[D]/dt. (c) Sketch curves for [A], [B] [C] and [D] vs. time. (d) The value of k2 was found to be 0.05 sec-1. Calculate the value of k-2. a) The equilibrium constants are gotten from the equilibrium concentrations: K1 = [C]/[A][B] = (1 × 10-11 M)/(6 × 10-10 M)2 = 2.78 × 107 M-1. K2 = [D]/[C] = (3.9 × 10-10 M)/(1 × 10-11 M) = 39.0. (I kept the units for the first constant in the biochemistry style.) b) d [C ] = k1[ A][ B ] − k −1[C ] − k 2 [C ] + k − 2 [ D ] = k1[ A][ B ] + k − 2 [ D ] − ( k −1 + k 2 )[C ] and dt d[ D] = k 2 [C ] − k − 2 [ D ] . dt concentration (nM) c) The first reaction is very fast, so it will come to equilibrium first. Then the second will slowly come to equilibrium. We know the starting concentrations. A and B will rapidly drop as the first reaction equilibrates and [C] increases. Then [D] will slowly increase to its final value as [C] drops. The first reaction will react to the change in [C] with drops in [A] and [B] to their final values. The 1 initial plateau when the second reaction 0.8 hasn’t yet kicked in will have [A] = [B] = 1 A nM – x and [C] = x: [C]/[A][B] = x/(1 nM and 0.6 B 2 7 -1 – x) = 2.78 × 10 M . If we expect x to 0.4 be small, then x = 2.78 × 10-11 M. Thus, D 0.2 [A] = [B] = 9.72 × 10-10 M and [C] = 2.78 × 10-11 M at the intermediate point. A C 0 0 0.2 0.4 0.6 0.8 1 sketch might look like time (relative to equlib) d) We know K2 which equals k2/k-2. So k-2 = k2/K2 = (0.05 s-1)/(39) = 1.28 × 10-3 s-1. Chapter 8 Problems Page 5 of 6 11/1/2007 8.21. A carboxypeptidase was found to have KM = 2 μM and kcat = 150 s-1 for substrate A. (a) What is the initial rate of reaction for [A] = 5 μM and [E]0 = 0.01 μM? (b) The presence of 5 mM of a competitive inhibitor decreased the initial rate by a factor of 2. What is the value of KI? (c) A competing substrate B is added to part (a). Its KM = 10 μM and kcat = 100 s-1. Calculate vB/vA. a) We need Vmax: Vmax = kcat[E]T = (150 s-1)(0.01 μM) = 1.5 μM/s. So plug into the M-M equation: V [ S ] (1.5 × 10 −6 M / s )(5μM ) = v0 = max = 1.07 × 10 − 6 M / s K M + [S ] 2 μM + 5μM b) A competitive inhibitor makes the apparent KM change, so first calculate what the apparent KM is: v0 = Vmax [ S ] (1.5 × 10 −6 M / s )(5μM ) 1.07 × 10 −6 M / s or KM,app = 9.02 μM. The = = K M + [S ] K M , app + 5μM 2 ⎛ [I ] ⎞ ⎟ or, after a little algebra, apparent KM in the presence of I is K M , app = K M ⎜⎜1 + K I ⎟⎠ ⎝ [ I ]K M (5mM )(2 μM ) KI = = = 1.42mM . K M , app − K M 9.02μM − 2μM c) The problem as stated seems to ask for two different things: i) the relative rates if the substrates are both present simultaneously at 5 μM and ii) the relative rates of hydrolysis if A and B are considered separately in separate reaction. The second is easier to deal with, so I do it first 1) We already have v0 for [A] = 5 μM from part a). Calculating for [B] = 5 μM gives Vmax, B [ S ] (1.0 × 10 −6 M / s )(5μM ) v0, B = = = 0.333 × 10 − 6 M / s . K M , B + [S ] 10 μM + 5μM Therefore, v0, B v0, A = 0.333 × 10 −6 M / s 1.07 × 10 − 6 M / s = 0.311 Notice that this is not what you get if you apply equation 8.25 since that equation is valid only if [A] and [B] are very small compared to their respective Km values (if the reactions are in separate pots). However, as we will see, if both substrates are present together in the same solution, then Eq. 8.25 does hold. 2) If both A and B are present together in the same solution, then simple MichaelisMenten analysis doesn’t hold. The reason is that, as far as A is concerned, some of the enzyme that it can interact with is removed by the EB complex. We need to account for this in the mathematics that leads to the expression for [EA], which gives us the velocity of consumption of substrate A. This is done exactly the same way that it is done when a competitive inhibitor I is present in the reaction mix. You can read about that on pages Chapter 8 Problems Page 6 of 6 11/1/2007 416-417 in the book. The whole “trick” is to note that the total amount of enzyme present, [ET], equals the sum of [E], [EA] and [EB], so that [EA] = [ET] – [E] – [EB] Now, we know that [ EB] = Menten, setting 1 [ E ][ B] (gotten in the same way as standard MichaelisKB d [ EB] = 0 = k1, B [ E ][ B] − k −1, B [ EB] − k 2, B [ EB] ). So dt [EA] = [ET] – [E] – [EB] = [ET] –[E] – ⎛ 1 [ B] ⎞ ⎟ [ E ][ B] = [ ET ] − [ E ]⎜⎜1 + KB K B ⎟⎠ ⎝ If you put this into our derivation of the Michaelis-Menten equation, you will get vA = k cat , A [ ET ][ A] ⎛ [ B] K M , A ⎜1 + ⎜ KM ,B ⎝ ⎞ ⎟ + [ A] ⎟ ⎠ Following the same logic for substrate B, you get v B = k cat , B [ ET ][ B] ⎛ [ A] ⎞⎟ K M , B ⎜1 + + [ B] ⎜ KM ,A ⎟ ⎝ ⎠ . Taking the ratio of vB to vA gives ⎧⎪ ⎛ k cat , B [ ET ][ B]⎨ K M , A ⎜1 + ⎜ ⎪⎩ vB ⎝ = vA ⎧⎪ ⎛ k cat , A [ ET ][ A]⎨ K M , B ⎜1 + ⎜ ⎪⎩ ⎝ ⎫ k cat , B ⎞ ⎧⎪ ⎟ + [ A]⎪⎬ [ B]⎨1 + ⎟ ⎪⎩ ⎪⎭ K M , B ⎠ = ⎧⎪ ⎫⎪ k cat , A [ A] ⎞⎟ [ A]⎨1 + + [ B]⎬ K M , A ⎟⎠ ⎪⎩ ⎪⎭ K M , A [ B] KM ,B [ B] [ A] ⎪⎫ k cat , B + [ B] ⎬ K M , B K M , A ⎪⎭ K M , B = [ A] [ B] ⎪⎫ k cat , A [ A] + ⎬ K M , A K M , B ⎪⎭ K M , A This is exactly equation 8.25, and it is valid no matter how small or large are the concentrations of A and B. The catalytic efficiencies for substrates A and B are, respectively, kcat,A/KM,A = (150 s-1)/(2 μM) = 75 s-1μM-1 and , kcat,B/KM,B = (100 s-1)/(10 μM) = 10 s-1μM-1. So clearly the enzyme has a strong preference for A over B. When the two substrates are at equal concentration, we have that the ratio of velocities vB/vA equals the ratio of the catalytic efficiencies, so vB/vA = (10 s-1μM-1)/(75 s-1μM-1) = 0.133. This holds when the two substrates are mixed together with the enzyme.