choline - Springer Static Content Server
advertisement

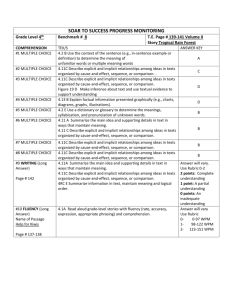
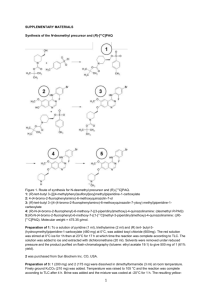
IMPD preparation example 1: [11C]choline Important notice: [11C]choline is a well established radiopharmaceutical, whose preparation was first reported 30 years ago. Since that time, several methods of preparation/purification have been published and adopted by the various involved Small Scale Radiopharmacies. Thus, [11C]choline may be prepared via [11C]CH3I obtained via the classic, liquid phase route (by reduction of [11C]CO2 in THF, addition of HI and subsequent distillation and transfer of [11C]CH3I into the dimethylaminoethanol precursor) or via the more recent “gas phase” route (with initial formation of [ 11C]CH4, and subsequent radical reaction with iodine). Even for the methylation reaction and subsequent purification, the methods may be different. Finally, every applicant could use different instrumentation (automated synthesis module, cyclotron, target, quality control equipment). The proposed example cannot account, for obvious reasons, for all of the above mentioned methods and techniques. It includes information and experimental data related to one of the possible preparation routes only. It is of paramount importance to remember that, during the preparation of the IMPD, every applicant should include the specific description of their own instrumentation, radiosynthetic pathway, experimental conditions, methods, data, etc. and also define their specifications with an appropriate justification. 2.2.1.S DRUG SUBSTANCE 2.2.1.S.1.1 Nomenclature Drug substance IUPAC name: ethanaminium, 2-hydroxy-N,N,N-[11C]trimethylammonium chloride CAS: (2-hydroxyethyl)-[11C]trimethylammonium chloride Synonims: [11C]choline, [methyl-11C]choline, [11C]trimethylethanolamine Radionuclide: C-11 2.2.1.S.1.2 Structure Fig. 1 – Structure of [11C]choline Molecular formula: C411CH14ClNO Molecular weight: 137.62 Stereochemistry: the active substance does not contain chiral carbon atoms CAS # 67-48-1 (referred to as choline chloride) 2.2.1.S.1.3 General Properties Physico-chemical properties: [methyl-11C]choline structure includes an atom of the positron emitting radionuclide C-11, whose characteristics are depicted in Table 1. 11 C decays to the stable isotope 11B, through the emission of β+ (99,8%), with a physical half-life of 20,38 min. 1 Parent Nuclide 11 C 6 T1/2 Decay mode 20.38 min β+ + Emax Relative intensity 960.2 KeV 99.759% γKeV Daughter nuclide 511 11 B 5 Table 1 – C-11 decay scheme (data from F. Ajzenberg-Selove, Nuclear Physics A506,1 (1990 - http://atom.kaeri.re.kr/cgi-bin/decay?C-11%20EC) Physicochemical characteristics of [12C]choline chloride Appearance White solid crystals, hygroscopic Solubility in water 650 g/L Solubility in other media Highly soluble in methanol, ethanol LogP -3.77 Melting point 244/247°C pH Choline chloride, at the typical working concentration, forms neutral, or close to neutral solutions, in water (pH = 6-7) LD50 in rats (i.v. injection) 53 mg/Kg Table 2 – List of selected physicochemical properties of “cold” choline. As stated in the above Table 2 header, the listed properties refer to the “cold”, non radioactive [12C]choline. As the difference with [11C]choline is only attributable to the replacement of a C-11 atom with a C-12 atom (choline chloride), the physicochemical properties reported in table 2 may also refer to the active substance [11C]choline. Due to its inherent radioactive nature, most of the typical molecular structural characterization test (e.g. MS, NMR) are not applicable with the radioactive compound. 2.2.1.S.2.1 Manufacturer(s) As stated above, the preparation of [11C]choline is usually a continuous process, and the active substance is, as a rule, not isolated. Thus, the information related to the manufacturer will be given in section 2.2.1.P.3.1. 2.2.1.S.2.2 Description of Manufacturing Process and Process Controls Due to the high emission energy of 11C, combined with the need to use considerable amounts of starting activity (typically >37 GBq) and the very short half-life of the above radionuclide, [methyl-11C]choline, as well as most 11C labelled radiopharmaceuticals, is prepared using fully automated radiosynthesis modules. They are capable to perform all the necessary operations, from the transfer of the radionuclide from the cyclotron, to the final formulation as an injectable solution of the radiopharmaceutical. For these reasons, these radiopharmaceutical preparations are considered as continuous processes carried out in closed systems. As a consequence, the active substance, as well as intermediates or by-products, are as a rule not isolated. Radionuclide production 11 C is generated “in-target” in the form of [11C]CO2 by means of an accelerated cyclotron proton beam via the nuclear reaction: 2 14 N(p,)11C The beam, with a projectile energy usually of 18 MeV, is directed on a target loaded with a gaseous mixture of 14N2 (99,5%) + O2 (0,5%). 11C “in-target” forming atoms react with the available oxygen molecules, to yield [11C]CO2, representing a typical example of the so called “hot chemistry”. Radionuclide production is fully automated. Typical irradiation conditions are strictly depending on the actual need and they are defined on a case by case basis. However, beam current for clinically useful amount of [11C]choline are generally in the range 30-40 A, while irradiation times are often in the range 40-60 min. The radionuclide production is carried out in a target made of aluminium with a volume of 50 cm3. Radiosynthesis of the intermediate [11C]CH3I (gas phase method) [11C]choline, as well as numerous 11C labelled radiopharmaceuticals, is synthesized via the formation of the useful intermediate [11C]CH3I, that may be prepared following two different radiochemical pathways, depending on the physicochemical form of the reactants. Indeed, they are defined as “gas phase” or “liquid phase (or wet)” chemistry pathways. [11C]CH3I production via “wet chemistry” method will not be described in the present document. In the [11C]CH3I “gas phase” preparation, the cyclotron produced [11C]CO2, purified and concentrated, is mixed with hydrogen (H2) and then delivered to a column loaded with a suitable nickel catalyst, at the temperature of 300-350°C, with formation of [11C]CH4. Water is then removed by passing the gaseous mixture through a column, downstream to the catalyst, loaded with P2O5 ascarite (NaOH, adsorbed on a silica pellet, 20-30 mesh); the latter plays the double role of efficiently absorbing the generated moisture and the unreacted [11C]CO2, as well. [11C]CH4 is then purified and concentrated by absorption on a suitable inert support (e.g. Carbosphere®), cooled to low temperature (e.g. -175°C) using e.g. liquid nitrogen. Heating the column, [11C]CH4 is rapidly released and transferred to a quartz tube, at 100°C, where it is mixed with sublimated I2, under a helium stream; the two reactants are then forced to re-circulate through a loop, which include a furnace previously heated at 720°C, where [11C]CH3I is formed. The reaction mechanism is as follows. I2 I + 11CH3 H 11 CH3 + I2 2I 11 CH3 + HI CH3I The by-product HI is absorbed by an additional ascarite column, while [11C]CH3I is concentrated, at room temperature, on a column loaded with a suitable polymeric matrix (e.g. Porapak® 50-80 mesh, Waters). As the radical reaction sequence above described is not very efficient, a single passage through the furnace is usually not sufficient to produce [11C]CH3I with high yield. To overcome this restriction, the reactants are forced to re-circulate through the loop several times, until the maximum [11C]CH3I activity is reached, by means of a suitable, low pressure, gas pump. [11C]CH3I activity is monitored by a radioactivity sensor, located in close proximity to the solid support column itself. At the end of the [11C]CH3I formation process, the above column is heated to 200°C, thus allowing the release and transfer of the 3 desired intermediate, under a helium stream, to the reaction vial, previously loaded with the DMAE precursor. Radiosynthesis and purification of [11C]choline Reaction vial has to be previously loaded with the DMAE precursor dissolved in acetonitrile. The methylation reaction takes place at 70°C for 3 min. The reaction schematic is as follows: Fig. 2 – Radiosynthesis of [11C]choline At the end of the above reaction, the mixture is purified through a cation exchange column, which selectively trap [11C]choline. The desired product is then eluted using an adequate volume of saline physiological solution, sterilized through a 0,22 μm filter, and collected in a sterile, pyrogen free glass vial. With the aim to avoid overpressure into the collection vial, a second, vented 0,22 μm filter should be inserted through the rubber closure. The overall synthesis time is 30 min. The radiochemical yield, calculated as the ratio between the final [11C]choline and the [11C]CH3I activity, is in the range 2535%, corrected for decay. The radiochemical yield is not determined by taking the starting [11C]CO2 or [11C]CH4 as the references, as their activities are difficult to quantitate with the radiosynthesis module, due to the extremely variable geometry of the above gases when they are trapped by the respective columns. As already stated above, the [11C]choline radiosynthesis, purification and formulation is an automated, continuous process, carried out in a closed system (the “synthesis module”). For this reasons, the active principle [11C]choline, as well as other intermediates, are, as a rule, not isolated. The synthesis module may be considered as a closed system because the starting materials (reagents, solvents, precursor), the intermediates and the final product, including the purification subset, are never exposed to the external atmosphere in any step of the whole process. 2.2.1.S.2.3 Control of Materials For most of the starting materials, controls are limited to a visual inspection, to verify the integrity of the packaging and products, and a check on the analysis certificate and expiry date. The most critical starting material is certainly the DMAE precursor. Moreover, it has to be underlined that in case of “gas phase” method, several starting materials might be re-used several times, and they are not replaced at the end of every synthesis. Examples are represented by I2, the nickel catalyst, the solid support used to trap volatile by-products and/or reagents, such as Carbosphere, Porapak, ascarite or P2O5. They are indeed replaced every 10 synthesis or more, depending on their stability and the rate of contamination. In Table 3 a list of the starting materials used in the preparation of 11 [ C]choline via “gas phase” method using a GE Tracerlab FxC-Pro, together with the proposed verification test, is reported. 4 Materials Dimethylaminoethanol (DMAE) [11C]CO2 P2O5 N-14 + 0.1-1% O2 Test and acceptance criteria HPLC; purity should conform with the indication of the supplier, based on analysis certificate C-11 has T1/2 = 20.3 min; this starting material cannot be isolated and tested during the radiosynthetic process See the attached analysis certificate See the attached analysis certificate; additionally, an irradiation test is performed every time the gas cylinder containing the gaseous mixture N2 + O2 is replaced See the attached analysis certificate See the attached analysis certificate See the attached analysis certificate See the attached analysis certificate Water for injection EtOH Iodine NaCl 0,9% injectable solution CH3CN See the attached analysis certificate Ni catalyst See the attached analysis certificate H2 See the attached analysis certificate Carbosphere See the attached analysis certificate Porapak See the attached analysis certificate Accel CM cartridge See the attached analysis certificate (SepPak) Acrodisc 0,2 µm filter, See the attached analysis certificate Supor membrane Sterile vials See the attached analysis certificate Vented Millex GS 0,22 µ See the attached analysis certificate filter Table 3 – List of the starting materials used in the preparation of [11C]choline via “gas phase” method using a GE Tracerlab FxC-Pro. 2.2.1.S.2.4 Control of Critical Steps and Intermediates The “in-process” controls are limited to the monitoring of the critical parameters (e.g. activity, reaction temperatures and pressures, inert gas flow rate) through the graphical control software interface. Printouts of representative preparation process diagrams are usually provided. A full quality control program is set for the final product (see section # 2.2.1.P.4.2.) 2.2.1.S.2.5 Process Validation and/or Evaluation Please refer to the section 2.2.1.P.3.5 2.2.1.S.2.6. Manufacturing Process Development Please refer to the section 2.2.1.P.2.3 2.1.2.S.3 Characterisation: 2.1.2.S.3.1 Elucidation of Structure and other Characteristics [12C]choline is provided with a suitable CoA, which states that NMR and MS spectra have been performed to confirm the choline structure. No further structure elucidating analyses are requested. With the aim to confirm the purity, an analytical HPLC control of the reference standard is performed. The experimental conditions are given in the section # 2.2.1.P.4.2. 5 2.1.2.S.3.2 Impurities Radionuclidic purity: Several European Pharmacopoeia Monographs of 11C labelled radiopharmaceuticals have been published to date. Acceptance criteria for radionuclidic purity for [11C]methionine, [11C]raclopride, [11C]flumazenil, [11C]acetate is usually set to > 99% of 11C, with maximum admitted amount of other radioisotopic contaminants being no more than 1%. As the radionuclidic impurities are generated during the cyclotron irradiation process, and considering that there are no significant differences between the [11C]choline preparation procedure and the above cited 11C radiolabelled compound, the same acceptance criteria has been adopted for the interested radiopharmaceutical. As for the possible radioisotopic contaminants, in addition to the main nuclear reaction 14N(p,)11C, there are other nuclear reactions that may take place at the cyclotron target level. The most prominent are 14N(p,n)14O and 16O(p,)13N. Other impurities arising from the irradiation of target foils (vacuum foil and target foil) are usually not of concern in case of gaseous target products such as [11C]CO2, as they do not transfer from the solid foils to the gaseous phase, and they are thus not swept out by the target during the unloading process. 13 N and 14O are both positron emitting radionuclides, and their presence cannot thus be detected through the gamma emission spectra analysis. On the other hand, 14O may be ruled out as a significant contaminant, considering its ultra-short half-life (T1/2 = 70.6 sec), combined with an average elapsed time between the end of bombardment (EOB) and the end of the synthesis (EOS) of 15-30 min, which means a number of decays in the range 12-25, more than enough to remove any trace of the above impurity. The only radionuclidic contaminant potentially present in the final product at End of Synthesis (EOS) is thus 13N, in the form of gaseous 13N labelled oxides (e.g. [13N]NO, [13N]NO2, etc.). Decay profile analysis may be conducted (see also section # 2.2.1.P.5.1 and Ph. Eur. monograph N. 0125, “Radiopharmaceuticals”) by measuring the activity of a radiopharmaceutical sample at three time points (T0, T1 and T2), using a suitable, calibrated instrument (e.g. dose calibrator, gamma spectrometry). Specification for 11C half-life, as reported in the Ph. Eur. chapt. N. 50700 (“Table of physical characteristics of radionuclides”) is 20.385 min. The above cited Ph. Eur. monographs for 11C labelled radiopharmaceuticals usually report an acceptance criteria in the range 19.9-20.9 min (i.e. ±2.5%), which has been adopted in the present document. Radiochemical purity The acceptance criteria for radiochemical purity might be, as already seen for radionuclidic purity, defined on the basis of the relevant published Ph. Eur. monographs for other 11C labelled radiopharmaceuticals, and set to > 95% of [11C]choline. Analytical method for radiochemical purity determination is analytical HPLC with integrated radiochemical detector. More details are given in section 2.2.1.P.5.2. Chemical purity The more plausible chemical impurities in [11C]choline preparations, are: i) the DMAE precursor, and ii) residual solvents such as acetonitrile, which is typically the reaction solvent, and ethanol/acetone, which are used for radiosynthesis module cleaning purposes. As neither dedicated Ph. Eur. monograph is available, nor the other 11C labelled radiopharmaceutical relevant monographs are of help in this case, the acceptance limit for the precursor DMAE has been established on the scientific literature and toxicity data basis (see for instance the paper “Doses as high as 1200 mg daily produce no serious side effects; and single 2500 mg dose taken in suicide attempt had no adverse effects”. [Gosselin, R.E., H.C. Hodge, R.P. Smith, and M.N. 6 Gleason. Clinical Toxicology of Commercial Products. 4th ed. Baltimore: Williams and Wilkins, 1976., p. II-240]), which indicate how the amount of precursor that could be likely present in the final preparation, even considering the worst case where all the DMAE used in the labelling reaction is detected in the finished product vial, is not of concern from the toxicity point of view, and negligible risks are thus associated. As for the residual solvents, their limits are defined in the document “EMEA note for guidance on impurities: residual solvents” (CPMP/ICH/283/95), and in chapter 5.4 of Ph. Eur. The analysis is usually performed using gas-chromatography. More details are given in the section 2.2.1.P.5.2. 2.2.1.S.4 Control of the Drug Substance: Details on methods for [11C]choline analysis, their validation, the batch analysis, and the justification of specifications will be provided in the appropriate 2.1.P sub-sections 2.2.1.S.5 Reference Standards or Materials: The list of reference standards is provided in Table 4. There are two distinct kind of reference standard: i) chemical standard, ii) radionuclide calibrated sources. As for the chemical standard, it has to be noted that a choline bitartrate standard has been added to the list. Indeed, the high hygroscopicity of the true standard choline chloride, make it difficult to handle and accurately weigh, thus decreasing the reliability of repeatability and linearity test. The chemical reference standards are commercially available, chemical grade products. The specifications for purity have been set by the supplier, and accepted by the Applicant. The specifications, and most of the analytical test (e.g. 1 H-NMR and/or 13C-NMR, mass spectrometry) are performed by the supplier, and described in the attached Certificate of Analysis. The chemical reference standards are re-tested for chemical purity by the applicant using HPLC, as detailed in the section 2.2.1.P.5. The radioactive reference standards are used to verify the calibration status of the gamma spectrometer and the dose calibrator, respectively. Their composition and the activity(ies) of the radionuclide(s) at reference time and date are described in the attached Certificate of Analysis. The calibrated source are metrologically referable to recognized standards, and a verification of their identity or purity is in this case non applicable Reference standard Choline bitartrate Choline chloride Multinuclide source Mononuclide source Aim Choline identity Choline identity Gamma spectrometer calibration Test HPLC HPLC Not applicabile Not Dose calibrator applicabile Table 4 - List of reference standards Acceptance criteria Purity > 95% Purity > 95% Not applicabile Not applicabile 2.2.1.S.6 Container Closure System For the reasons already stated above, container closure system will be described for the drug product, in the appropriate sections. 2.2.1.S.7 Stability For the reasons already stated above, stability will be described for the drug product, in the appropriate sections. 7 2.2.1.P INVESTIGATIONAL MEDICINAL PRODUCT UNDER TEST 2.2.1.P.1 Description and Composition of the Investigational Medicinal Product: Each vial contains 15 mL of a saline physiological solution of [11C]choline, with radioactive concentration in the range 250-3700 GBq/mL, at reference time. Reference time is taken as the time when the activity contained in the finished drug product is measured with the dose calibrator. The [11C]choline solution is contained in 20 mL borosilicate glass vials (Ph. Eur. type I), sealed with bromobutyl rubber stopper (Ph. Eur. compliant), which is in turn secured with an aluminum flip-off cap. Composition The composition of the finished drug product is given in Table 5: Component Quantity Function Reference [ C]choline 370-3700 MBq/mL Active substance N.A. NaCl 0,9% solution 14-16 mL Excipient Ph. Eur. 11 Table 5 – Composition of the injectable solution of [ C]choline 11 2.2.1.P.2 Pharmaceutical Development [11C]choline is prepared using an automated synthesis module, by means of which the desired product is simply purified, at the end of the labelling reaction, via the well known “solid phase extraction” technique, using cation exchange cartridge, which selectively retains [11C]choline. After washing the cartridge with water, [11C]choline is then eluted with saline physiological solution. The final formulation is thus very simple and NaCl 0.9% is the only excipient included in the product vial. The active substance is never isolated in the course of the preparation process, and it is available for quality controls only at the end of formulation step. The finished product has to be considered as a multi-dose preparation, and the activity at reference time may vary, depending on the clinical trial need (e.g. the higher the number of daily subjects, the higher the number of individual radiopharmaceutical doses that should be prepared). Stability studies have been performed, demonstrating that the composition of the [11C]choline solution does not undergo significant changes to chemical and radiochemical purity during the considered time (2 hours). For more information about stability, see section 2.2.1.P.8. 2.2.1.P.2.3 Manufacturing Process Development Not applicable 2.2.1.P.3 Manufacture: 2.2.1.P.3.1 Manufacturer(s) Institution Name Address Person responsible for the small scale preparation of radiopharmaceuticals e-mail address Phone Fax 2.2.1.P.3.2 Batch Formula A batch of [11C]choline is usually represented by a single, multi-dose vial. The materials used in the preparation of a typical batch via the “gas phase” method and 8 using a GE Tracerlab FxC-Pro radiosynthesis module are listed in Table 6. The starting materials marked by an asterisk are not single use, and they are replaced with a frequency established during the automated device validation procedures. Starting materials Amounts DMAE 0,025 mL *P2O5 2g Water for injections (WFI) 10 mL *Ascarite 15 g *Iodine 3,5 g NaCl 0,9% physiological solution 15 mL CH3CN 0,475 mL *Ni Catalyst 200 mg H2 50 mL/min *Carbosphere 450 mg *Molecular sieves 300 mg SepPak Accell cartridge 1 Table 6 – List of starting materials used in a [11C]choline batch production via “gas-phase” Batches of [11C]choline to be used in clinical trials are typically made of a single vial. 2.2.1.P.3.3 Description of Manufacturing Process and Process Controls The [11C]choline preparation process has already been described in section 2.2.1.S.2.2 of the present document. However, a flow chart is added below: 9 Fig. 3: flowchart of the preparation process of [methyl-11C]choline 2.2.1.P.3.4 Controls of Critical Steps and Intermediates Description of in-process controls and intermediated has already been given in section 2.2.1.S.2.4 2.2.1.P.3.5 Process Validation and/or Evaluation Process validation has been performed by preparing and controlling three consecutive batches of [11C]choline. Validation runs have been performed in the same operating conditions (e.g. starting [11C]CO2 activity, amount of precursor, physicochemical reaction parameters) characteristic of the process; i.e. using the same instrumentation and starting materials in the stated quantities normally set for typical runs. The parameters evaluated and their acceptance criteria are found in Table 7: 10 Parameters Acceptance criteria Radioactive concentration 0.37 - 3.7 GBq/mL Final volume 15 1 mL Table 7 – Production parameters evaluated during the process validation The experimental data are reported in the following table: [11C]choline activity Radioactive concentration Volume Conform BATCH 1 BATCH 2 BATCH 3 (Prod. (Prod. (Prod. 29/09/10) 07/10/10) 08/10/10) 10,7 GBq 8,4 GBq 10,22 GBq 0,713 GBq/mL 0,56 GBq/mL 0,681 GBq/mL 15 mL 15 mL 15 mL □ yes □ No □ yes □ No □ yes □ No Table 8 – Experimental production data for process validation Each batch has been analyzed following the complete quality control program. The results are summarized in Table 9. Specifications are described in section 2.2.1.P.5.1. 11 QUALITY CONTROL Batch 1 Acceptance Test Batch 2 Batch 3 Confor results Conform results Conform results 4.5-8.5 6.5 □ Yes □ No 6.22 □ Yes □ No 6.47 Clear, colourless ≤ 4.1 mg/Vmax* ≤ 50 mg/Vmax* 0.1 mg/Vmax* Clear, colourless 4.37 g/mL 414 g/mL 0 □ Yes □ No □ Yes □ No Clear, colourless □ Yes □ No 0 mg □ Yes □ No 570 g/mL □ Yes □ No Clear, colourless 1.38 g/mL 287 g/mL 0 □ Yes □ No 0 100% □ Yes □ No 100% □ Yes □ No 100% 506 KeV □ Yes □ No 505 KeV □ Yes □ No 505 KeV 99.9% 99.9% □ Yes □ No 99.9% □ Yes □ No 99.9% Half-life 19.9-20.9 min 20.35 min □ Yes □ No 20.6 min □ Yes □ No 20.2 min Bubble point ≥ 46 psig 48 psig □ Yes □ No 55 psig □ Yes □ No 48 psig □ Yes □ No Sterile □ Yes □ No Sterile □ Yes □ No Sterile □ Yes □ No <5 EU/mL □ Yes □ No <5 EU/mL □ Yes □ No < 5 EU/mL □ Yes □ No criteria pH Appearance CH3CN EtOH DMAE Radiochemical purity Identification: gamma spect. Radionuclidic purity Sterility Bacterial endotoxins ≥ 95% of [11C]choline 511 10 KeV Ph. Eur. Conform Ph. Eur. Conform (<175/Vmax* in IU/mL) □ Yes □ No □ Yes □ No Table 9 – Experimental quality control data for process validation *= max. recommended dose (mL) 2.2.1.P.4 Control of Excipients: 2.2.1.P.4.1 Specifications The excipients used in the preparation process of the finished investigational radiopharmaceutical should meet the specifications reported in Table 10. Excipient Specification 0.9% NaCl Ph. Eur. Table 10 – List of excipients The 0.9% NaCl solution is approved following a check for packaging integrity, expiry and Certificate of Analysis. The appearance should also be verified. For the above reasons, for excipient control there is no need to describe analytical procedures, as well as their validation and justification of specification(s). No novel excipients have been used. 2.2.1.P.5 Control of the Investigational Medicinal Product: 2.2.1.P.5.1 Specifications Each batch of [11C]choline is submitted to quality control, with the aim to evaluate chemical, radiochemical, radionuclidic and biological purity of the finished product. The QC tests are summarized in Table 11. 12 m □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No □ Yes □ No Parameters Appearance Test Visual inspection Identification HPLC Identification Gamma Spectrometry Radiochemical purity DMAE Chemical Purity EtOH* CH3CN* Radionuclidic purity* Half-life pH Bacterial endotoxins* Filter integrity Sterility* HPLC HPLC GC GC Gamma spectrometry Gamma spectrometry/ Dose calibrator pH meter Specification Clear, colourless solution The principal peak in the radiochromatogram obtained with the test solution of [11C]choline has approximately the same retention time as the principal peak in the chromatogram obtained with a reference solution of “cold” choline chloride (or choline bitartrate) Gamma photons have an energy of 511 KeV and, depending on the measurement geometry, a sum peak of 1022 KeV may be observed [11C]choline ≥95% ≤0.1 mg/max. recommended dose (mL) ≤50 mg/max. recommended dose (mL) ≤4.1 mg/max. recommended dose (mL) 11C ≥ 99 % 19.9-20.9 min 4,5-8,5 Ph. Eur. <175 EU/ max. recommended dose (mL) Bubble point Following specification of the supplier Ph. Eur. Sterile Table 11 – Specifications and acceptance criteria for [11C]choline *The tests labelled with an asterisk are performed after the release of the radiopharmaceutical, due to both the test duration not compatible with radionuclide half-life (e.g. sterility test or radionuclidic purity), and/or for radiation protection reasons (e.g. bacterial endotoxins test). 2.2.1.P.5.2 Analytical Procedures Analytical procedures are described in detail in relevant Standard Operating Procedures, which are available on request. 2.2.1.P.5.2.1 Identification and determination of the chemical (DMAE) and radiochemical purity using HPLC [11C]choline analysis: specifications - Identification: the main radiochemical peak should have the same retention time evidenced by the SST test for standard choline chloride (or bitartrate) ±0.2 min - Radiochemical purity: peak area for [11C]choline has to be > 95% of the total peak areas - Chemical purity (DMAE): the peak area for DMAE should not be more than the corresponding area of the peak obtained with reference SST solution 13 2.2.1.P.5.2.2 Determination of residual solvents using gas-chromatography Instrumentation: Gas-chromatograph Head space injection system FID-Detector (Flame Ionization Detector) Capillary column: 30m, 0.32 mm, 1 µm, Acquisition data system Carrier Gas: helium Operative conditions: Temperature ramp: 0-80°C at 10°C/min, etc. Flow: 1mL/min Acquisition time: 10min [11C]choline analysis: specification - Acetonitrile: maximum admitted concentration = 4.1 mg/V, where V is the maximum recommended dose (mL). - Ethanol: maximum admitted concentration = 50 mg/V, where V is the maximum recommended dose (mL). 2.2.1.P.5.2.3 Gamma spectrometry Instrumentation: Gamma Spectrometer NaI detector Acquisition data software Identification Method: the activity of the [11C]choline sample should be in a suitable range, such that the instrument dead time is < 5%. Analyze the sample for a suitable time (e.g. 10 min), and energy spectrum (e.g. 0-1800 KeV). Specification: the only gamma photons have an energy of 0.511 ± 10% MeV and, depending on the measurement geometry, a sum peak of 1.022 MeV may be observed. Radionuclidic Purity Method: a sample of [11C]choline with suitable volume and geometry is left to decay for a time sufficient to allow the complete decay of the main radionuclide (11C). Usually this decay time should be estimated such as the remaining 11C activity should be negligible (e.g. Tdecay >20 half-lives). Analyze the sample for a suitable time (e.g. 60 min), and energy spectrum (e.g. 0-1800 KeV). Specifications: the sum of the areas due to the possible contaminants, recalculated to the activity at shelf-life of the radiopharmaceutical preparation, and related to the total radioactivity of the preparation itself, should be <1% of the total peak areas. 2.2.1.P.5.2.4 Determination of the half-life Instrumentation: Gamma Spectrometry NaI detector Acquisition data software 14 Or Dose calibrator Method: Decay analysis should be performed by measuring the sample at least two times (T 0 and T1), with a suitable interval between T0 and T1 (e.g. 10 min). [11C]choline: specifications Half-life of [11C]choline should be in the range 19.9-20.9 min. Calculations may be done using the decay equation: where: - t = time between the two activity measurements N0 = activity at T0 N = activity at T1 2.2.1.P.5.2.5 Determination of pH Instrumentation: pH meter - Before to proceed with the analysis, a calibration test using suitable reference buffer solutions has to be performed. - Insert the pHmeter electrode into the sample solution, and record the value [11C]choline analysis: specification pH of the test solution should be in the range 4.5-8.5 2.2.1.P.5.2.6 Endotoxins Instrumentation: Endosafe PTS Reader [11C]choline analysis: specifications Bacterial endotoxins level should be <175 EU/V, where V is the maximum recommended dose (mL). 2.2.1.P.5.2.7 Filter Integrity Test This test is required if the finished product is sterilized using an 0.22 m filter membrane. Instrumentation: - Remotely operated device specifically designed for filter integrity test Method - Connect a suitable gas source upstream to the filter membrane - Gradually increase the pressure - Record the pressure value at which bubble formation is observed [11C]choline: specifications Bubble point depends on the filter characteristics, and the specifications are given by the manufacturer. 2.2.1.P.5.3 Validation of Analytical Procedures The reference guidelines are as follows: 15 ICH Harmonised Tripartite Guideline “Text on validation of analytical Procedures” Step 4 of the ICH Process, November 2005 ICH Harmonised Tripartite Guideline “Validation of Analytical Procedures: Text and Methodology” Step 4 of the ICH Process, November 2005 The above guidelines may not always apply to validation of radioactive compounds, due to their peculiar nature. Exceptions will be discussed. 2.2.1.P.5.3.1 Method validation for the determination of chemical purity using HPLC Validation of analytical method for the determination of chemical purity of injectable solution of [11C]choline is here presented. In Table 12 the validation parameters and their acceptance criteria are summarized: Acceptance criteria Test ≥ 2,5 Specificity CV % 2% Repeatability Fcalc≤ value of Ftab Intermediate precision R2 ≥ 0,99 Linearity Quantification Limit (LOQ) Limit of detection (LOD) CV % 2% for standard choline CV % 5% for impurity --Fcalc≤ value of Ftab Robustness Table 12 – Test and acceptance criteria for the validation of the method for the determination of chemical purity using HPLC Specificity Specificity determination is performed analyzing mixture containing critical components that might be present in the finished product [11C]choline solution, and demonstrating that the method is capable to distinguish the various components present at the limit concentration for the considered standards. The [11C]choline preparation method development did not prompt for chemical impurities, except for the DMAE precursor. Thus, analyses were performed using a series of solution containing the precursor itself, together with the choline chloride (or bitartrate) standard. Peak resolution may be calculated using the following equation: Rs 1,18 Trb Tra Wa Wb where: Trb= retention time of the compound b Tra = retention time of the compound a Wb= full width at half-height of the compound b Wa= full width at half-height of the compound a 16 Linearity The statistical function used in these cases is linear regression with least squares. The curve equation, the correlation coefficient and the determination coefficient (r 2) are then calculated. Equation: y = ax + b where: a = slope b = intercept y = peak area x = analyte concentration r2 should be 0,99, within the concentration operating range. The above evaluation requires the preparation of at least 5 standard solutions with different concentrations for each of the interested analytes (DMAE and standard choline). Such solutions are usually prepared by a series of dilution starting from a “mother” solution, with the highest concentration. Precision Precision may be considered at different levels, as a measure of repeatability or intermediate precision. a. Repeatability: Repeatability may be calculated based on the content of standard DMAE and choline. The statistical parameter of concern is the variation coefficient (CV%) (or Relative Standard Deviation. RSD), which is determined using the following equation: CV % s 100 m where: s = standard deviation of the peak areas m = average of the peak areas CV% should be < 2% for the standard choline, and < 5% for the DMAE precursor. The necessary experimental data may be obtained by injecting 6 times a sample of the desired analyte, whose concentration should fall within the range established during linearity test. b. Intermediate precision Intermediate precision may be determined through the variance calculation (ANOVA), which allows, in turn, for Fisher value calculation. Limit of quantitation (LOQ) Experimentally, LOQ may be determined by analyzing a series of diluted solutions of DMAE and standard choline, until a concentration level quantified with a precision >95% is reached. The experimental value determined as above described need to be confirmed through a precision analysis, using a sample at the concentration corresponding to the found LOQ. Acceptance criteria is CV% <5%. Limit of detection (LOD) The LOD may be determined experimentally by successive dilutions, until the above minimum concentration is found. Robustness In case of HPLC analysis, a critical parameter might be the mobile phase flow. Once the critical parameter has been selected, three consecutive analyses of the desired analyte have to be performed, following a deliberate modification of the pump flow. 17 Data may be evaluated through the variance analysis (ANOVA). In the present document, the parameters whose variation will be analyzed are retention time and peak areas. 2.2.1.P.5.3.2 Method validation for the determination of radiochemical purity using HPLC Validation of the analytical method for the determination of the radiochemical purity is here presented. In Table 13, the validation parameters and their acceptance criteria are summarized: Test Specificity Repeatability Intermediate precision Linearity Acceptance criteria Not applicable CV % 2% Not applicable R2 ≥ 0,99 Limit of quantitation (LOQ) Not applicable Limit of detection (LOD) Not applicable Robustness Not applicable Table 13 – Test and acceptance criteria for the validation of the method for the determination of radiochemical purity using HPLC In case of validation of methods of radioactive compounds, some of the ICH guidelines validation parameters may not be of concern and do not apply. In particular: - Specificity would require the analysis of at least two radioactive analytes with comparable activity, and this is usually not applicable in case of very short half-life radionuclides labelled compounds, for experimental reasons. - Flow cell radiochemical detectors typically used in the analysis of radiolabelled compounds are inherently not very suitable for quantitation purposes; moreover, they are typically used to determine relative ratios between the activity of the various labelled compounds that might be present in the sample, rather than for true activity quantitation purposes, and the final required outcome is a percentage areas calculation. Thus, both LOQ and LOD analyses have been considered of no concern. - Similar considerations may be done for intermediate precision analysis, which is strictly related to the possibility of quantitation of the desired analytes. - As for the robustness, the results obtained during the tests for validation of chemical purity, using “cold” analytes (DMAE and standard choline), provide response and data that may apply to the radioactive analytes, as well. A 18 repetition of the test would in this case represent a useless radiation burden for the operators. - Last but not least, the ALARA (“as low as reasonably achievable”)” radiation protection concept should indeed always be kept in mind, while designing the necessary validation tests. Linearity Considering the radioactive nature and the very short half-life of 11C, the typical experimental approach, based on the preparation of a series of solution with different concentrations does not apply. On the contrary, in this case one sample solution only, with a suitable radioactive concentration, is analyzed 5 times, at defined time intervals. Indeed, the radioactivity being the physical parameter of concern for radiochemical detectors, the radionuclide decay itself provide the necessary linear series of values. r2 may thus be extrapolated from the calibration curve by analyzing 5 different radioactive concentration of [11C]choline. Repeatability Here also apply the same considerations described for linearity; that’s, the decay of the radionuclide 11C inevitably lead to a decrease in time of the radioactivity. However, repeatability may be evaluated analyzing a series of HPLC runs obtained with repetitive injections of a single [11C]choline sample, and recalculating the obtained peak area values with the decay equation: lnA0= ln A + λt λ= 0,693/t1/2 where: A0= corrected peak area A= measured peak area t= time interval between the considered injection and the first one t1/2= half-life (11C = 20,3 min) The peak area values, normalized for decay, may then be compared and yield a consistent statistical analysis. Average, standard deviation and (CV%) are then calculated. Repeatability has to be determined in three different days, to verify the instrument outcome during the time course. 2.2.1.P.5.3.3 Validation of the analytical method for the determination of residual solvent using gas-chromatography Gas-chromatography is used to evaluate the amount of residual solvents in the finished product solution of [11C]choline. The validation parameters are practically the same already described for the validation of the method for the determination of chemical purity (see Table 12, section 2.2.1.P.5.3.1). For this reason, discussion will not be repeat in this context. 2.2.1.P.5.3.4 Validation of the analytical method for the determination of radionuclidic purity and identification test using gamma spectrometry Validation parameters are described in Table 14: 19 Test Specificity Repeatability Intermediate precision Acceptance criteria Rs ≥ 1 CV % 2% Not applicable Linearity R2 ≥ 0,99 Accuracy 90-110% Limito of quantitation (LOQ) Limito of detection (LOD) Robustness Not applicable Not applicable; MDA* Not applicable Table 14 – Test and acceptance criteria for the validation of the method for the determination of radionuclidic purity and identification test using gamma spectrometry *LOD may be suitably replaced by minimum detectable activity (MDA) values, which are determined by the instrument every time a sample is measured. MDA is a parameter depending by several factors such as geometry, activity, background, counting time, etc. It is assumed that all the intended measurements are performed keeping into account the above factors, so as to obtain consistent data. As already discussed in section 2.2.1.P.5.3.2, validation parameters stated by ICH guidelines are not always applicable when dealing with radioactive samples, essentially due to inherent decay phenomena. Thus, intermediate precision and robustness may not be evaluated with sufficient reliability, and the remaining validation parameters need to be adapted to the peculiar nature of the radioactive samples. LOD may be considered, provided that the instrument is periodically calibrated for efficiency. In the case of robustness, it is not easy to apply deliberate and controlled variations to the normal operating detector parameters, and for this reason this part of the validation program is not considered. Specificity Specificity may be determined using a suitable, preferably multi-nuclide, calibrated source, with an overall emission energy spectrum capable to include as much as possible the instrument operating range (usually 0-2000 KeV). For this reason, [11C]choline samples are not useful in this case, as their emission energy is likely to be attributable to the expected 511 KeV only, and they usually do not contain significant and detectable amounts of radioisotopic impurities. Thus, the specificity analysis may just provide a general validation of the inherent physical characteristic of the instrument, more than a true evaluation of the [11C]choline injectable solutions. Acceptance criteria for specificity (Rs) should take account of the above considerations and, in particular, of the differences in resolution between NaI and HPGe (or other similar high resolution detectors). Indeed, the former have a lower resolution and Rs values should necessarily be lower, as well. For calculation purposes, the following formula has been used: 20 Rs 1,18 Erb Era FWHM a FWHM b where: Erb= Energy at the centroid of the radionuclide b peak Era = Energy at the centroid of the radionuclide a peak Wb = full width at half-height of radionuclide b peak Wa = full width at half-height of radionuclide a peak Linearity As the desired analyte is [11C]choline, whose activity decrease with time, the same consideration already discussed in section 2.2.1.P.5.3.2 apply. Repeatability The same set of data obtained following the linearity tests may be used to determine repeatability, as already described in section 2.2.1.P.5.3.2. Accuracy Accuracy has been evaluated using a multinuclide calibrated source containing 60Co, which has been selected, among the others, due to its long half-life (T1/2 = 5.27 y) and its widespread availability. Accuracy has been determined by comparing calculated activity of 60Co with the activity quantified by the instrument. 2.2.1.P.5.3.5 Validation of analytical method for the determination of pH As the pH meter and related methods are very simple, validation following the ICH guidelines is not necessary. The only suggested test is to check the repeatability of the analytical method. CV% may be determined by analyzing 6 times a series of suitable buffer solutions, whose pH brackets a significant portion of the pH working range (e.g. buffer solutions with pH of 4.7 and 10, respectively). Anyhow, the instrument calibration is checked every time it has to be used, using the same buffer solutions above depicted. 2.2.1.P.5.4 Batch Analyses Data related to the [11C]choline batches are included in the respective certificates of analysis. They report information related to the number and batch size, as well as information on the production site, methods of production, quality control and acceptance criteria. 2.2.1.P.5.5 Characterization of Impurities The discussion about the possible impurities in injectable solution of the finished product [11C]choline has already been described in section 2.2.1.S.3.2, related to the active substance. 2.2.1.P.5.6 Justification of Specification(s) As already mentioned in the appropriate sections, the analytical test, the methods and acceptance criteria have been derived, where applicable, from the general Ph. Eur. monograph “Radiopharmaceuticals” and from the Ph. Eur. monographs specifically dedicated to 11C labelled radiopharmaceuticals above cited. It is noteworthy that the radiopharmaceutical [11C]choline is a very well established radiopharmaceutical, for which a comprehensive scientific and clinical literature is available, that cover all the aspects related to the preparation and quality control of the radiopharmaceutical and, most noticeably, to its clinical outcome. Thus, the specifications adopted in the present document may be considered as a suitable summary of numerous experiences and propose a preparation process already validated, in practice, by several production sites. 21 2.2.1.P.6 Reference Standards or Materials Reference standards and materials have already been discussed in section 2.2.1.S.5. 2.2.1.P.7 Container Closure System The radiopharmaceutical product is contained in a glass vial, Ph. Eur. type I, sterile and pyrogen free, covered with a bromo (or chloro) butyl rubber stopper (Ph. Eur. conform), sealed with a flip-off aluminum cap. Attached are the certificate of analyses of the vials and stopper manufacturer. No further test other than visual inspection is applied to these materials. 2.2.1.P.8 Stability The radiopharmaceutical [11C]choline, due to the short half-life of 11C, is typically of immediate use, and its administration to patients immediately follows the preparation and quality control operations. Generally speaking, stability does not need to be determined in case of very-short half-life radionuclides such as 11C. However, a stability study has been performed, with the aim to provide evidence on how the quality of [11C]choline may vary with time and to establish a shelf life for the finished product, under recommended storage conditions. During the whole shelf-life, the radiopharmaceutical characteristics of purity have to meet quality criteria discussed and established in the previous sections. The goal is thus to define expiry time for the radiopharmaceutical product. For stability study purposes, three consecutive batches of [11C]choline have to be prepared by using the maximum possible starting activity of the radionuclide 11C, in order to obtain batches with the highest radioactive concentration, that allow to evaluate the effect of radiolysis of the active substance in “worst case” conditions. For each batch of radiopharmaceuticals, the following information should be provided: - batch number - preparation date and time - calibration time - activity at calibration time - radioactive concentration at calibration time Specifications and acceptance criteria for each batch are those defined in section 2.2.1.P.5.1, Table 11. The test program follows the matrix listed below: 22 Test Appearance Identification: HPLC Identification: gamma spectrometry Radiochemical purity DMAE Chemical Purity EtOH CH3CN Radionuclidic purity* Half-life pH Bacterial endotoxins Filter integrity Sterility** Specification Clear, colourless solution The principal peak in the radiochromatogram obtained with the test solution of [11C]choline has approximately the same retention time as the principal peak in the chromatogram obtained with a reference solution of “cold” choline chloride (or choline bitartrate) Gamma photons have an energy of 511 KeV and, depending on the measurement geometry, a sum peak of 1022 KeV may be observed [11C]choline ≥95% ≤0.1 mg/max. recommended dose (mL) ≤50 mg/ max. recommended dose (mL) ≤4.1 mg/ max. recommended dose (mL) 11C ≥ 99,9 % T0 X T1 X T2 X X / / X / / X X X / X / X / / X / / X / / X / X X X X / X / X / / / / / 19.9-20.9 min 4,5-8,5 <175 EU/ max. recommended dose (mL) Following specification of the supplier Sterile Table 16 – Matrix of the test to be performed during stability study (“X” means: to be tested; “/” means: not to be tested) *The test has to be performed at least 12 h after the batch preparation, to allow the complete decay of the main radionuclide 11C **Sterility test is performed after adequate decay of the radioactivity contained in the finished product For example, in case the proposed shelf-life is 2 h, the analyses will have to be repeated three times, at the following time intervals: T 0 (EOS), T1 (T0 + 60 min) and T2 (T0 + 120 min).). Considering the very short half-life of 11C, and the typical amounts of the above radionuclides that may reasonably be produced using commercially available cyclotrons and target systems, it seems to be reasonable to propose a shelf-life no longer than 2 h. 23
![ACCURACY OF [11C] CHOLINE POSITRON EMISSION](http://s3.studylib.net/store/data/006910188_1-178035aba028502f62a71ecfd059e7d4-300x300.png)

![[125I] -Bungarotoxin binding](http://s3.studylib.net/store/data/007746915_2-24484224552cb6e93390ec53e82e3abc-300x300.png)