Supplementary Information (doc 52K)
advertisement
")
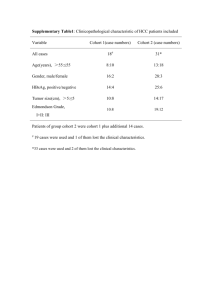
Supplementary Information Supplementary methods CNV analysis Sample preparation We used a CNV early access array for CNV analysis, which is designed based on the database of known CNVs. Each sample and reference DNA (3.0 μg each) was labeled with Cy5 or Cy3, respectively, using the Agilent DNA labeling kit (Agilent Technologies, Inc., Santa Clara, CA, USA). Following the manufacturer’s recommended hybridization and washes, the arrays were scanned with an Agilent MicroArray Scanner G2505A and the obtained TIFF image data were processed with Agilent Feature Extraction software (version 9.5.3.1) using the CGH-v4_95_Feb07 protocol. CNV early access array data analysis Extracted data were analyzed with Agilent DNA Analytics 4.0 software (version 4.0.85), and the Aberration Detection Method 2 (ADM-2) algorithm 1 was used to identify contiguous genomic regions that corresponded to chromosomal aberrations. The following parameters were used in this analysis: threshold of ADM-2: 6.0; centralization: on (threshold: 6.0, bin size: 10); fuzzy zero: off; aberration filters: on (minProbes = 2 AND minAvgAbsLogRatio = 0.5 AND maxAberrations = 10000 AND percentPenetrance = 0); feature level filters: on (gIsSaturated = true OR rIsSaturated = true OR gIsFeatNonUnifOL = true OR rIsFeatNon- UnifOL = true). At a minimum, two contiguous suprathreshold probes were required to define a change. To find an obvious homozygous deletion, aberrant regions with a signal log ratio of less than –5.0 were 1 searched. Genomic positions were based on the UCSC March 2006 human reference sequence (hg18) (NCBI build 36.1 reference sequence assembly). To find copy number differences between the twins, we detected their respective copy number changes compared to a reference. Then we calculated the fold change of each probe in the regions and selected the probes with fold changes of more than 1.2. Real-time PCR analysis We performed real-time PCR analysis using SYBR-green dye (Applied Biosystems, Foster City, CA, USA) with an ABI PRISM 7900HT (Applied Biosystems) to confirm the copy number differences between the twins detected by arrays. For real-time PCR analysis, we selected as candidates those regions that contained several probes showing consecutive changes in the same direction of fold change or that contained a probe showing large absolute values of fold change. In total, we selected 40 regions and one immunoglobulin-related region was included as a positive control for the experiment. Apolipoprotein B (APOB) was used as a single control gene, and the copy numbers in the candidate regions were calculated as a relative ratio to APOB. For quality control, a gene on the X chromosome (X inactive-specific transcript, XIST) was also examined using an unrelated female sample. Applied primer sets were shown in Supplementary Table 2. Reference S1 Lipson, D., Aumann, Y., Ben-Dor, A., Linial, N. & Yakhini, Z. Efficient calculation of interval scores for DNA copy number data analysis. J Comput Biol 2006; 13: 215-228. 2 Supplementary Figure Legends Supplementary Figure 1. Copy number profiles of CNV array in pair 1 (a) and pair 2 (b). Copy number profiles in sex chromosome reflect the effect of female reference sample. The copy number profiles within each twin pair were nearly identical. Supplementary Figure 2. The chromosomal distribution of copy number differences in pair 1(a) and pair 2(b). Each bar shows the number of probes for which fold changes were more than 1.2 (FC > 1.2) in each chromosome. In both twin pairs, marked differences were restricted to chromosomes 2, 14, and 22, which contain immunoglobulin (Ig)-related regions. Supplementary Figure 3. Results of real-time PCR analysis. (a) A difference in copy number between pair 1 twins in the Ig-related region on chromosome 22 was detected by CNV array. Any other copy number differences detected by CNV arrays between the twin pairs were not confirmed by real time PCR. (b) The detected CNV was confirmed by real-time PCR analysis (FC = 2.12). (c) The copy number differences of XIST on the X chromosome in male twins and a control female. The result ensures the quality of this experiment (FC = 2.02 for healthy co-twin, 1.92 for bipolar twin, respectively). Supplementary Figure 4. The flowchart of tiling array data analysis and detection of candidate regions for case-control analysis. Bold numbers represent the number of MRs selected in each step. The filtering process was described in detail in supplementary materials and methods. In brief, 1) the data of each twin pair were directly compared 3 and the regions (containing 6 or more CpG sites) showing significant differences (p value < 10-4) between the BD twin and the healthy co-twin were selected. The selected MRs were named BD (bipolar twin)-dominant MRs and C (healthy co-twin)-dominant MRs. 2) The data of each twin were compared with those of a reference sample (i.e., unmethylated DNA). Among the BD-dominant MRs, the regions showing the significantly methylated signal compared with a reference sample in BD twin but not in healthy co-twin were selected. The selected regions were named BD-specific MRs. C-specific MRs were determined vice versa. 3) The MRs overlapping with a CpG islands were selected. 4) Based on the results of bisulfite sequencing of representative regions, we applied a more stringent threshold for filtering (p value < 10-6) for the direct comparison between each twin pair. 5) The regions that showed alteration of DNA methylation status before and after the transformation by Epstein–Barr virus in our previous study using 4 sets of lymphoblastoid cell lines (LCLs) and peripheral blood cells were excluded. 6) From the candidate regions for BD-specific MRs, the regions that were detected as MRs in at least one of 4 LCLs were excluded. From the C-specific MRs, only the regions overlapping with common MRs in all of 4 LCLs were selected. Supplementary Figure 5. Results of bisulfite sequencing of the 13 candidate regions. Regions in which the differential DNA methylation was not confirmed (No) had p value greater than 10−6. The regions that were partially confirmed (Partial) also had p value around 10−6. Regions where methylation was completely confirmed (Complete) had p value less than 10−6. Note that partially confirmed or non-confirmed results were attributable to the false negative signals from tiling array (see Supplementary Fig 6). We did not observe false-positive results at the examined sites. 4 Supplementary Figure 6. The representative results of bisulfite sequencing. Among the 13 regions, differences in DNA methylation in 11 regions were confirmed (5 regions were completely confirmed, 6 regions were partially confirmed) and differences in 2 regions were not confirmed (Fig. S5). Examples of a completely confirmed region (a), a partially confirmed region (b), and a region that was not confirmed (c). The results of tiling arrays in a pair of monozygotic twins are shown above. The vertical axis represents the signal intensity, and the horizontal axis represents the base number (NCBI36/hg18). The CpG island is shown by a gray square. The region showing statistically significant methylation difference between the bipolar twin and the healthy co-twin, and the region examined by bisulfate sequencing are shown by a black square and a red bar, respectively. The results of bisulfite sequencing are shown on the bottom. Black circles represent the methylated CpGs, and the white circles represent the unmethylated CpGs. Each row shows the data of one clone. Supplemntary Figure 7. Perspective view of the methylation difference between the twins in chr17. The vertical axis represents the signal intensity, and the horizontal axis represents the base number on the chromosome 17 (NCBI36/hg18). The methylation of SLC6A4 promoter region was enlarged in a box. The region showing statistically significant methylation difference between the bipolar twin and the healthy co-twin is shown by a red bar. Supplementary Figure 8. DNA methylation difference between twins in FANK1. a) Results of comprehensive DNA methylation analysis of LCLs of a pair of 5 monozygotic twins discordant for BD using tiling arrays. The vertical axis represents the signal intensity, and the horizontal axis represents the base number on the chromosome 10 (NCBI36/hg18). Exon-intron structure of the FANK1 is shown below the data of tiling arrays. The CpG island is shown by a gray square. The region showing statistically significant methylation difference between the bipolar twin and the healthy co-twin, and the region examined by bisulfate sequencing are shown by a black square and a red bar, respectively. b) Results of bisulfite sequencing. The genomic region detected statistically significant methylation difference between the twins by tiling arrays, which corresponds to the base numbers from 127573680 and 127574304, is shown above. The five CpG sites are shown by red letters with under bars. Black and white circles represent the methylated and unmethylated CpGs, respectively. Each raw shows the data of one clone. Five circles in one raw represent the five CpG sites shown above. This region is equally methylated in both twins. Supplementary Figure 9. DNA methylation difference between twins in KIAA1530. a) Results of comprehensive DNA methylation analysis of LCLs of a pair of monozygotic twins discordant for BD using tiling arrays. The vertical axis represents the signal intensity, and the horizontal axis represents the base number on the chromosome 4 (NCBI36/hg18). Exon-intron structure of the KIAA1530 is shown below the data of tiling arrays. The CpG island is shown by a gray square. The region showing statistically significant methylation difference between the bipolar twin and the healthy co-twin, and the region examined by bisulfate sequencing are shown by a black square and a red bar, respectively. 6 b) Results of bisulfite sequencing. The genomic region detected statistically significant methylation difference between the twins by tiling arrays, which corresponds to the base numbers from 1353373 and 1354215, is shown above. The 22 CpG sites are shown by red fonts with under bars. Black and white circles represent the methylated and unmethylated CpGs, respectively. Each raw shows the data of one clone. Five circles in one raw represent the five CpG sites shown above. This region is hypermethylated in both twins. 7