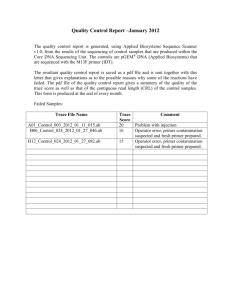
sequence amplification
advertisement

Molecular Biology-2015 1 Manipulating sequences 1. There are several different manners by which a sequence can be presented. In this exercise we will examine these. Let’s start with a relatively simple short sequence: 5’- GAATGCGGCTTAGACTGGTACGATGGAAC-3’ A different version of the above sequence would be to present its complement. Write the complement of the above sequence. Note that your sequence must be presented in the 3’>5’ orientation. 2. Another way of presenting a sequence is referred to as the inverse (or reverse) sequence. This involves simply presenting the sequence in the opposite orientation. Rewrite the sequence you generated in step 1 in the 5’>3’ orientation. 3. The final way of presenting a sequence is what is called the reverse complement. Relative to the initial sequence presented in step 1, the sequence you generated in step 2 represents the reverse complement. 4. This can also be achieved with a computer program such as the web based program “Reverse complement”. You can access this program from the link on this course’s web site under the heading bioinfo links. Obtain the reverse sequence of the template and print it for your assignment. Obtain the complement sequence of the template and print it for your assignment. Obtain the reverse complement sequence of the template and print it for your assignment. Obtain the reverse sequence of the complement sequence of the template and print it for your assignment. Obtain the complement sequence of the reverse sequence of the template and print it for your assignment. Molecular Biology-2015 2 Predicting PCR amplification products One of the projects of this lab is to amplify, mutagenize and clone the E.coli lacZ gene from the vector pUC19. To accomplish this, primers were designed to specifically amplify this gene. Primer alignment using Blast 1. To determine which region will be amplified we will perform a sequence alignment. To do so two different approaches will be used. The first one will involve using Blast at the NCBI web site. 2. Go to Blast and select the option "Align two or more sequences" below the query box. A new Blast program window will be displayed as shown below: 3. Obtain the sequence of the vector pUC19 from this course’s web site from the sequence menu and copy paste it in FASTA format in the first query box. 4. Copy-paste the sequence, in FASTA format, of the first forward primer we will be using to amplify the LacZ sequence in the second query box (Page 33 of the lab manual: PCRmutagenGAT). 5. Choose the option “somewhat similar sequences” as you've done in a previous exercise. Molecular Biology-2015 3 6. Now click on Blast to obtain your results. Scroll down the page to view the alignment as shown below: This display shows the alignment of the query sequence (the sequence entered in the first box), which represents your template sequence, with that of the subject sequence (the sequence entered in the second box), which represents the primer sequence. A great deal of information can be obtained from this display: i. ii. iii. iv. First, note that in the above example the alignment start point is at base 7 of the subject sequence, meaning that the first 6 bases did not match. Second, a perfect match was found between bases 668-691 of the query sequence. Amongst the aligned bases, 24 out of 24 were identical (See identities = 24/24). A perfect match was obtained between the two sequences as entered (See Strand = Plus/Plus). This means that if the subject sequence were that of a primer, its sequence is identical to that of the query sequence. It would therefore NOT anneal to this strand, but rather to the reverse complement of this strand. Molecular Biology-2015 4 7. Record the position on the query sequence (template strand) of the alignment. You want the smaller of the two values. 8. Now repeat this exercise with the reverse primer (PCRmutagenRev). You will notice this time that the alignment is: Strand=Plus/Minus. The reason for this is that this second primer sequence is the same to the reverse complement strand and should therefore anneal to the opposite strand. 9. Obtain the position of the alignment. This time you want the greater value. You should now be able to predict the size of the amplified region and say which primer is “Forward” Strand=Plus/Plus and which one is “Reverse” Strand=Plus/Minus. In order to obtain the length of the amplification product subtract the alignment position of the second primer from that of the first. Larger number – smaller number. 10. Repeat this exercise with the next Forward primer; GCCTGCAGGTCGACTCTCGAGGAACC (PCRmutagenGAA). Note from the alignment that a mismatch was detected. Obtain the position on the template for the mismatch. Indicate it in your assignment as per the following example: C118→G. Primer alignment using Clustal Omega 1. Another program which can be used to align primer sequences to a template sequence is Clustal Omega. Use the link on this course's web site under the heading Bioinfo links to access this program. You should obtain the following page: 2. This program allows you to align either protein or nucleotide sequences. Since we are interested in aligning nucleotide sequences change the sequence parameter to "DNA" 3. Then, enter your sequences, the pUC19 and PCRmutagenGAT sequences, in FASTA format, in the query box. Note all sequences, must be entered in the same box and separated by a line break. Also, the sequence identifiers must be sufficiently different. See the example on the next page. Molecular Biology-2015 5 Sequence identifiers Line break 4. Change the output format to Clustal w\ numbers. 5. Now click on submit. Once the analysis is completed the alignment will be shown on a new page similar to the one shown below. 6. In contrast to Blast, a global alignment is shown rather than only the aligned sequence. Perfect matches are indicated by a star, whereas mismatched bases are indicated by a symbol other than a star or no symbol at all. In the above example two mismatches were detected, one of which represents the 3' base. Note that this would not have been shown using Blast and may therefore be more difficult to notice. The base on the 3' end is one of the most important. Primers which have a mismatch at this position will NOT work. 7. In contrast to Blast, this program performs the alignment only of the entered sequence and not on the reverse complement strand. To illustrate this, repeat the above steps with the pUC19 and the PCRmutagenRev sequences. You will notice this time that the alignment found is much less perfect. 8. Consequently, when aligning primers using Clustal omega, you should always verify with which template strand one obtains the best alignment. To determine whether the alignment is better on the other strand replace the original pUC19 sequence used with its reverse complement. Use the web based program "reverse complement" previously used to convert the sequence and then repeat this exercise. Typically, if the best alignment is obtained with the original sequence the primer is said to be a forward primer. If the best alignment is obtained with the reverse complement strand of the template, the primer is said to be a reverse primer. Molecular Biology-2015 6 Primer mapping Map the alignment positions of each of the following primer sequence on the pUC19 template sequence. A. B. C. D. TGCGGTGTGAAATACCCT GCCATTCAGGCTGCGCAA GGGTTATTGTCTCATGAG GAGACAATAACCCTGATA Use a diagram, as shown below, to indicate the annealing positions of each of the primers. Each primer should be indicated by an arrow; where the head of the arrow represents the 3' end and the tail represents the 5' end. The direction of the arrow should indicate whether the primer is a forward (→) or a reverse (←) primer. The position should represent the template position corresponding to the 5' end of the primer. Indicate in a legend to your figure all primer pairs, if any, would give an amplification product of at least 200bp. 814 1514 pUC19 56 908 2351