AD_abstract_2007
advertisement

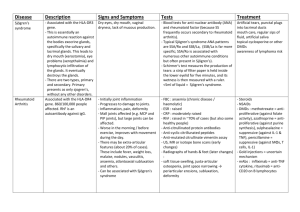
Autoimmune diseases Autoimmune diseases are diseases which are caused by immune reaction against selfantigens. Requirements: 1. evidence of autoimmune reaction (autoantibodies) 2. evidence of primary pathogenetic significance of this reaction 3. absence of another well-defined cause of the disease A spectrum of diseases may involve: 1. one single organ (IDDM, Hashimoto thyroiditis) 2. multisystem disease (SLE) 3. intermediate (Goodpasture syndrome) Mechanism of an autoimmune disease is the loss of self-tolerance. Self-tolerance : lack of responsiveness to an individual's own antigens Central (in thymus and bone marrow) and peripheral tolerance Mechanisms of peripheral tolerance : clonal deletion antigen sequestration clonal anergy peripheral suppression Mechanisms of autoimmune diseases : breakdown of T-cell anergy failure of activation-induced cell death failure of T-cell-mediated suppression molecular mimicry polyclonal lymphocytic activation release of sequestered antigens exposure of cryptic self and epitope spreading Genetic and microbial factors in autoimmunity. Systemic lupus erythematosus SLE is a multisystem inflammatory febrile disease, characterized by production of numerous autoantibodies ( particularly antinuclear ). It is a chronic remitting and relapsing disease with injury to skin, kidneys, serous membranes, joints, blood vessels and heart. Etiology The cause of SLE is unknown. Autoantibodies in SLE : 1. antibodies to plasma proteins (complement, clotting factors) 2. antibodies to cell surface antigens (blood cells) 3. antibodies to intracellular cytoplasmic components (RNA, ribosomes, lysosomes) 4. antibodies to nuclear antigens (DNA, histones, nucleolar antigen) Antibodies to double-stranded DNA and Smith (Sm) antigen (small nuclear ribonucleoprotein particle) are diagnostic of SLE. APLA – antiphospholipid antibodies are directed against plasma proteins that bind to phospholipids. In vitro APLA interfere with the clotting test (lupus anticoagulant), in vivo patients exhibit hypercoagulable state. Pathogenesis of SLE : 1. Genetic predisposition ( MHC class II , complement, additional unidentified genes ) 2. Environmental factors ( drugs, ultraviolet light, sex hormones ) 3. Immunologic factors Various factors lead to failure of regulation of self-tolerance, activation of T helper cells and B cells and production of numerous antibodies against nuclear and cytoplasmic components. The visceral lesions are caused by deposition of DNA-anti-DNA immune complexes ( hypersensitivity type III ) and direct cell toxicity ( hypersensitivity type II ). Pathology The most characteristic lesions result from the deposition of immune complexes and are found in the blood vessels, kidneys, connective tissue and skin. An acute necrotizing vasculitis of small arteries and arterioles may be present in any tissue. The arteritis is characterized by fibrinoid necrosis in the vessel walls; in chronic stage vessels undergo fibrous thickening. Skin : “ butterfly “ lesion ( erythema ) of face ( 50 % of patients ), urticaria, bullae, maculopapular lesion, ulcerations. Sunlight accentuates the erythema. Histology : liquefactive degeneration of the basal layer, edema at the dermal junction, edema and perivascular mononuclear infiltrates in the dermis. Joints : nonerosive synovitis without deformity. Acute phases – exudation of neutrophils and fibrin into the synovium and a perivascular mononuclear cell infiltration in the subsynovial tissue. CNS : noninflammatory occlusion of small vessels by intimal proliferation. Serosal cavities : acute, subacute or chronic fibrinous inflammation. Heart : in the majority of patients pericarditis. Myocarditis is less common. Nonbacterial verrucous endocarditis ( Libman – Sacks ) – vegetations on both surfaces of leaflets, single or multiple, irregular, 1 – 3 mm warty deposits. Coronary atherosclerosis ( in young patients with long-standing disease and treatment by corticosteroids ). Spleen : follicular hyperplasia, numerous plasma cells, and perivascular fibrosis of central penicilliary arteries ( onion – skin lesion ). Lungs : pleuritis and pleural effusion ( most common ), less common edema, hemorrhage and interstitial fibrosis. Chronic Discoid Lupus Erythematosus – skin manifestations of SLE, but systemic manifestations are rare. 5% to 10% of patients with discoid lupus develop multisystem disease over many years. 35% of patients have a positive ANAs test, but autoantibodies to doublestranded DNA and Sm antigen are not encountered. Subacute Cutaneous Lupus Erythematosus is intermediate between SLE and Discoid Lupus. It is characterized by papular lesions on the trunk. Drug-induced Lupus Erythematosus can occur following administration of procainamide, hydralazine, isoniazid, D-penicillamine. Unlike SLE, drug-induced lupus shows no sex predominance, patients are older than 50 years, renal and CNS involvement is rare. Unusual to find antibodies to double-stranded DNA AND Sm antigen. Autoantibodies to histones are typical of druf-induced lupus. The disease remits after withdrawal of the drug. Rheumatoid arthritis A chronic systemic inflammatory disease with symmetric and bilateral chronic polyarthritis Nonsupurrative proliferative synovitis which leads to destruction of joint cartilage and ankylosis of the joints Other organs involved : skin, heart, blood vessels, muscles, lungs RA is an autoimmune disease triggered by exposure of a genetically susceptible host to an unknown arthritogenic antigen. Pathogenesis : 1. the autoimmune reaction ( activated CD 4+ T cells and probably B cells ) 2. the mediators of joint injury 3. genetic susceptibility 4. antigens Rheumatoid factor is antibody IgM, sometimes IgG or IgA, directed against the Fc fragment of IgG. RF are also found in patients with SLE, scleroderma, and dermatomyositis. Pathology Joints initially the synovium is edematous, thickened and hyperplastic with bulbous fronds infiltration of synovial stroma by dense perivascular inflammatory infiltrates ( CD 4+ T cells, B cells, plasma cells, macrophages ) increased vascularity with hemosiderin deposits aggregation of fibrin covering the synovium and floating in the joint space (rice bodies) accumulation of neutrophils in the synovial fluid and not deep in the synovial stroma osteoclastic activity in underlying bone with formation of juxta-articular erosions, subchondral cysts and osteoporosis pannus formation – the pannus is inflammatory synovium, containing granulation tissue and fibroblasts, which grows over the articular cartilage and erodes it. After the destruction of cartilage the pannus bridges the apposing bones and forms a ankylosis ( fibrous and bony ). Inflammation in the tendons, ligaments and adjacent skeletal muscle frequently accompanies the arthritis Skin Rheumatoid nodules are the most common cutaneous lesions in RA. They arise in regions that are subjected to pressure ( forearm, elbows, lumbosacral area, legs ). They may appear in the lungs, spleen, pericardium, myocardium, heart valves, aorta. Rheumatoid nodules are firm, round to oval, large nodules may ulcerate. Microscopically, they have a central zone of fibrinoid necrosis with surrounding rim of macrophages, lymphocytes and plasma cells. Blood vessels Patients with severe erosive disease, rheumatoid nodules and high titers of rheumatoid factor are at risk of developing vasculitic syndromes. The involvement of medium to small arteries is similar to PAN. Obliterating endarteritis in small arteries results in peripheral neuropathy, ulcers and gangrene. Leukocytoclastic venulitis produces purpura, cutaneous ulcers and nail bed infarction. Lungs Chronic pleuritis, diffuse interstitial fibrosis, intrapulmonary rheumatoid nodules, pulmonary hypertension. Clinical course It is variable. Slowly beginning - more than 50% of patients, acute onset – 10%. Small bones of the hands and feet, wrists, ankles, elbows, and knees cervical spine are involved. The joints are swollen, warm, painful and stiff. The disease course may be slow or rapid, with the greatest damage during the first 4 -5 years. 20 % of patients enjoy periods of partial or complete remission. Life expectancy is reduced by mean of 3 to 7 years. Complications of RA are systemic amyloidosis, vasculitis and iatrogenic effects of therapy. Juvenile rheumatoid arthritis It begins before the age 16 and the arthritis must be present for a minimum duration of 6 weeks. JRA differs from RA ( oligoarthritis is more common, systemic onset is more frequent, large joints are affected more often, rheumatoid nodules and rheumatoid factor are usually absent ) Scleroderma ( progressive systemic sclerosis ) Autoimmune disease of connective tissue, characterized by vasculopathy and excessive collagen deposition in skin and internal organs ( lungs, GIT, heart, kidneys, etc. ) F : M = 3:1 25 – 50 years of age Familial incidence Association with HLA – DQB1 Pathogenesis The cause is not known. The trigger is a combination of abnormal immune responces and vascular damage, resulting in accumulation of growth factors that act on fibroblasts and stimulate collagen production. Inappropriate activation of humoral immunity: 1. hyperactivity of B – cells ( hypergammaglobulinemia and cryoglobulinemia ) 2. specific for SS antibodies (antibodies to Scl-70, anticentromere antibodies) Abnormalities of cellular immune system: T – cell activation ( CD4+ ) Releasing of cytokines and mediators ( IL-1, IL-2, IL-13, TNF, PDGF, TGF-β ) Activation of inflammatory cells and fibroblasts and excessive production of collagen. Microvascular disease presents early in SS: Intimal proliferation Endothelial injury Periadventitial fibrosis Ischemic injury and scarring. Morphology Skin Diffuse sclerotic atrophy of skin, begins in fingers and extends proximally. In early stage – edema, perivascular infiltrates with CD4+ T-cells, swelling and degeneration of collagen. Microvascular damage, partial occlusion. Progression – fibrosis of dermis, atrophy of epidermis, loss of appendages, calcifications. Advanced stage – cutaneous ulcerations and autoamputation ( fingers ), limitation of motion, face – drawn mask. GIT Progressive atrophy and fibrous replacement of muscularis. Esophagus – dysphagia, GERD, Barrett metaplasia. Small bowel – loss of villi and micrivilli – malabsorption. Musculoskeletal system Inflammation and hyperplasia of synovia, later fibrosis. Joint destruction is not common. Kidneys Early fibromuscular thickening of subintima luminal narrowing fibrosis In afferent arterioles – fibrinoid necrosis. Glomerular changes are not specific. Lungs Diffuse interstitial fibrosis end-stage pulmonary fibrosis ( honeycomb lung). Endothelial dysfunction pulmonary hypertension. Heart Patchy myocardial fibrosis. Congestive heart failure is uncommon. Clinical features: Diffuse scleroderma (progressive systemic sclerosis) and limited form (CREST variant). Progressive SS – severe and progressive disease of skin and early involvement of visceral organs. The CREST is a milder disease, characterized by calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly and telangiectasia. Involvement of viscera occurs later, and in general the patients live longer than those with SS. Inflammatory myopathies ( polymyositis and dermatomyositis ) Chronic inflammatory myopathies Rare autoimmune disease in children and adults Strong association with HLA-DR3, HLA-DRw52 and HLA-DQ Symmetric proximal painful muscle weakness, involvement of pharyngeal, neck-flexor and respiratory muscles Increased serum levels of muscle-derived enzymes Nonsuppurative inflammation of skeletal muscle Overlapping with other autoimmune diseases – SLE, scleroderma, rheumatoid disease, Sjogren syndrome Malignancy: association with bronchial, nasopharyngeal carcinoma The most common morphological characteristics: 1. presence of inflammatory cells 2. necrosis and phagocytosis of muscle fibers breast, ovarian, gastric and 3. mixture of regenerating and atrophic fibers 4. fibrosis Polymyositis occurs after the age 20 years, more common in females. Pathogenesis: direct muscle cell damage, produced by cytotoxic T-cells ( CD4+ ). Morphology: 1. endomysial inflammation ( CD8+ T-lymphocytes and macrophages ) 2. scattered degenerating or regenerating fibers 3. angiopathy is absent 4. perifascicular atrophy is not present. Dermatomyositis affects children and adults, more common in females. Pathogenesis: 1. immune complexes of IgG, IgM and complement in walls of capillaries 2. microangiopathy with loss of capillaries 3. injury and atrophy of myofibers 4. perivascular infiltrates of B and T-cells ( predominantly CD4+ ). Muscle injury is produced by complement-mediated cytotoxic antibodies directed against the microvasculature of skeletal muscle. The microangiopathy leads to ischemic injury of individual fibers and to fiber atrophy. Involvement of larger intramuscular arteries may lead to infarcts. The rash is related to microangiopathy. Extramuscular manifestation: interstitial lung diseases, vasculitis, myocarditis, involvement of GIT ( in juvenile dermatomyositis ). Morphology: 1. perivascular and perimysial lymphoid infiltrates ( B- cells and T –cells, predominantly CD4+ ) 2. intramuscular blood vessels – endothelial hyperplasia, fibrin thrombi, obliteration of capillaries 3. perifascicular atrophy 4. necrotic fibers and regeneration. Diagnosis: 1. clinical symptoms 2. EMG 3. elevated CK 4. biopsy – required for definitive diagnosis Sjögren syndrome Dry eyes (keratoconjuctivitis sicca) and dry mouth (xerostomia) are result of immunologically mediated destruction of lacrimal and salivary glands. Primary and secondary Ss. Middle-aged women, female-to-male ratio is 9:1. Etiology and pathogenesis The cause of Ss is unknown. Lymphocytic infiltration (CD4+ helper T cells and some B cells) and fibrosis of the lacrimal and salivary glands. 75% of patients have rheumatoid factor, 50-80% ANAs. SS-A(RO) and SS-B(La) are antibodies directed against ribonucleprotein antigens, can be detected in up to 90% of patients. Morphology and clinical features Periductal and perivascular lymphocytic infiltration in lacrimal and salivary glands, hyperplasia of ductal epithelium, atrophy of acini and ducti, fibrosis and replacement of the parenchyma with fat. D.D. with lymphoma. Complaints of patients : dry eyes, blurring of vision, burning, itching, atrophy and cracking of oral mucosa, xerostomia, involvement of bronchial, gastrointestinal glands. Extraglandular tissues (kidneys, lungs, blood vessels, nervous system) are involved in 25% of cases. Kidney – interstitial nephritis. 40-fold increased risk of malignant lymphoma. Treatment is symptomatic.