Protocol
advertisement

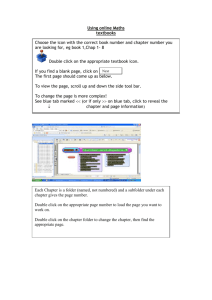
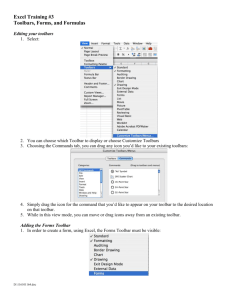
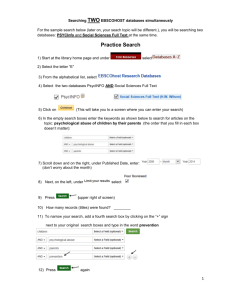
Rutgers Biomedical Engineering 125:315 Laboratory # 9: 2004 Cellular Property Measurements via Biomedical Imaging and Microscopy Developers: Becky Hughey (rhughey@eden.rutgers.edu) Eric Wallenstein (ejw42@eden.rutgers.edu) Faculty Instructors: Prof. P. Moghe (moghe@rci.rutgers.edu) Introduction: Lab Subsection 1: Cell Imaging: Viability Study Using Calcein and Ethidium Homodimer Many methods exist to visualize and quantify characteristics of cells. In this lab section cell viability will be studied. In every living cell there are general and unique enzymatic activities that define cell viability and function. Within viable cells, cytoplasmic esterases can cleave calcein-AM, resulting in the emission of a green wavelength signal when excited by a UV light source. However, if these enzymatic reactions are inhibited, i.e. following the introduction of toxins into the cell cytoplasm or through the permeation or degradation of internal membranes, necrosis (a type of cell death) will ensue. Furthermore, following cell membrane disruption or compromised cell integrity, another compound, ethidium homodimer-1, will have access to nucleic acids within the nuclei of these dead cells to produce a vibrant red fluorescent color. Lab Subsection 2-1: Image Quantification: Monitoring Cell Differentiation Depending on their overall health, activity, and environmental conditions, cells in culture/experimental regimes can display wide morphological ranges. For example, by applying sequential addition of growth factors, embryonic cells can be induced to express various differentiated cell types, each with unique cell shape characteristics. Differentiation can then be followed in such systems, through the classification of differentiated states based on measurable morphological characteristics. I. Purpose: Lab Subsection 1: Cell Imaging: In this lab, cell population viability will be assayed through the use of a calcein/ethidium homodimer stain. Live cells will stain green due to calcein binding, and dead cells will stain red due to ethidium homodimer binding to nucleic acids. Total cell population and percentage dead cells will be obtained using the Microsuite software package, quantifying on the parameter basis of color and cell diameter. Total cell population # cells expressing both ethidium homodimer and calcein Percentage dead cells # ethidium homodimer stained cells Total cell population Lab Subsection 2: Image Quantification: During this subsection, different morphological cell configurations will be quantified, compared, and related to function. In one system, differentiated embryonic stem cells will be quantitatively characterized based on major and minor cell axes measurements. The characterization scheme obtained from these measurements will then be used to classify cell populations as differentiated, or undifferentiated. II. Materials: Lab Subsection 1: Cell Imaging: The viability assay will be performed using previously acquired images of calcein/ethidium homodimer staining. Quantification for the viability assay will be performed using the Microsuite software package. Cell counts will be performed on the basis of color as well as cell size. Lab Subsection 2: Image Quantification: Quantification of cell phenotype and morphology will be performed with the Microsuite software package. III. Methods and Data Collection: Lab Subsection 1: You will be using cell imaging software to quantify the viability of a certain population of cells. The following procedure is used for counting the dead cells in the entire picture. The first image you will bring up and analyze contains cells previously stained with calcien and ethidium homodimer-1. These cells were imaged on the epifluorescence microscope in A209. Open Olympus MicroSuite software from the desktop. 1. From File toolbar, open <My Documents/lab6_microscopy/Images/live_dead> and open the DEAD image. This will open the image on the screen and place it in the image manager on the left side of the screen. The image manager allows you to keep many images at the ready and toggle between images. (i.e. you don’t have to open and work on one image at a time). Next, in step 2, you will create a new toolbar that will contain items we will refer you to in the course of the lab. 2. Open Special menu and select Edit Button Bars; Put a check next to Analysis and click OK. You will set color thresholds so that the program can select cells from the background 3. Go to the Measure menu and select Pixel Value 4. Click on two different cells and once on the black background 5. Right click and end the quantification process; Click Yes to stop measurements This will give you the red, green, and blue values of the pixel in chart form. These values will be used as starting points for color thresholding 6. On the analysis toolbar click the Set Color Threshold icon 7. Click the Window button in the Preview; move the box around and enclose a region with a good number of cells 8. Right click to lock the box in place 9. Select All in the Preview box 10. Put the representative minimum and maximum pixel values for each color obtained from the Pixel Values Sheet into the slider bar boxes for the representative color; Click OK 11. Adjust the values in the boxes so that the cells in the window become completely colored 12. Save the Pixel Value data sheet that you generated to the desktop to later e-mail to yourself The color thresholds are now set up and you can identify the cells from the background You will now set up a filter to sort the cells by diameter 13. Go to the Measure menu again and select Arbitrary Distance 14. Use this tool to measure the diameter of a cell 15. Repeat for two more cells 16. Right click and end the quantification process; Click Yes to stop imaging This will give you an idea of the range of diameters you will incorporate into your filter Next you will create a classification scheme to detect dead cells in the picture 17. On the analysis toolbar click on the Define Classification icon 18. Put the name DeadClass into the New Classification box and click New 19. Click on the Units button and select Pixel as Basic Unit. Pick Normal so that your units are pixels – a measure of distance. Click OK. Note: if you wanted to use area as your selection criterion, you would use pixels2 since these are units of area, not distance. 20. Click on the Compute button 21. Set min at the lowest value from the sheet obtained using Arbitrary Distance calculations 22. Set max at the highest value from the sheet obtained using Arbitrary Distance calculations 23. Set bins at 5 (this creates 5 categories of cell sizes from the values you entered) 24. Click OK to close the window and accept the scheme; Click OK on the Define Classification box. You will now specify which measurements we want computed for the items that are detected by your classification scheme. 25. On the analysis toolbar click on the Define Measurements icon 26. Select the particles tab. ID Class and ID Particle will already be selected 27. Select Area and Diameter Mean as measurements This will show you the mean diameter as well as the area of the cells you detect. 28. Click OK You will now specify the criterion you want to sort by (in this case you will pick diameter since you specified pixel and not pixel2 in the Define classification section) and using the subdivisions you set up (DeadClass as specified in the Define Classification menu). 29. On the analysis toolbar click on the Define Detection icon 30. Click on Detection Tab and make sure the ROI box is not checked. Instead select Frame This will allow you to analyze the entire picture. 31. Click on Classification tab 32. Select Diameter Mean as Criterion 33. Select DeadClass as Classification 34. Select Outline as the style 35. Select ID Particle as the Measurement label type 36. Click OK 37. On the analysis toolbar click on the Detect icon 38. On the analysis toolbar click on the Particle Results icon This will put all your results on a spreadsheet. Notice how various groups are outlined in different colors, corresponding to the values in DeadClass. You can also export to excel and remove data with no ID Class (not colored). Review your spreadsheet. How many particles that fit into your classifications where found. 39. Save the image of the measured picture, as well as the sheet of results obtained for Particle Results, not from ROI Results, to the desktop to later e-mail. 40. Now repeat this procedure (steps 3-39) to image live cells. Go to <My Documents/lab6_microscopy/Images/live_dead> and open the LIVE image. Lab Subsection 2: In the previous subsection, we quantified the morphology of all cells that fell within a certain region. In the next subsection we will manually select a few individual cells and measure their morphometric properties. Go to <My Documents/lab6_microscopy/Images/differentiation> and open the differentiated image Because this is not a fluorescent image you will have to reset your color thresholds. 1. On the analysis toolbar click the Set Color Threshold icon 2. In threshold slider boxes set minimum to 0 and maximum to 255 for all three colors. This will allow the program to analyze all cells You will now create regions of interest (ROI) around individual cells to analyze. 3. 4. 5. 6. 7. On the analysis toolbar click the Define ROI’s icon Select the Pencil Tool (freehand polygon). Left click and hold down to encircle a cell. Right click to close the outline Repeat this process (4-6) two more times This will define three ROI’s 8. Make sure the regions you created are selected and click close. This does not mean that your regions have been lost. They are still being stored somewhere in the program. You will now select the parameters to quantify. 9. On the analysis toolbar click on the Define Measurements icon 10. Click on the ROIs tab. 11. Select Area ROI, and Diameter Mean ROI. 12. Click OK. 13. On the analysis toolbar click on the Define Detection icon 14. On the Detection tab, select ROIs as the Search Area. 15. On the Classification tab, set Criterion to No Classification. 16. Click OK. 17. On the analysis toolbar click on the Detect icon 18. On the analysis toolbar click on the Frame ROI Results icon The ROI data is presented for the specified parameters, with the ROI listed as the row variable, and the parameter listed as the column variable. Save the spreadsheet (that which is accessed using the button for ROI results) and images of the selected cell populations on the desktop to later e-mail. Having learned two ways to calculate parameters of cells, quantify the following properties using a Classification method of your choice. 1. Maximum diameter 2. Elongation 3. Orientation You will analyze the image entitled Undifferentiated, found at <My Documents/lab6_microscopy/Images /differentiation> Save the image of the measured picture as well as the data sheet of results obtained using one of the above classification methods on the desktop to later e-mail. Lab section 3: To view the staining of cytoskeletal components in the cell we will make a composite image. Open the images from <My Documents/lab6_microscopy/Images/composite/Phalloidin> and <My Documents/lab6_microscopy/Images/composite/Phase> Here the image manager allows you to open both images at one time and toggle between them. 1. 2. 3. 4. 5. Find the Define Fluorochromes icon (painters pallet) in the upper toolbar and click Click deselect all Select <Phase Contrast> and FITC Click OK Go to the Define Fluorescences icon (next to Define Fluorochromes icon) 6. Put a check in the box for both <Phase contrast> and FITC (You may need to add FITC as a new target.) 7. Click OK 8. Highlight both images – click on one, then hold shift and click on the other 9. Go to Create Composite image icon (to the right of the Define Fluorescences icon) 10. Define the type of fluorescence for each image in the fluorescence drop down window; Phalloidin = FITC, Phase = Phase contrast 11. Put a check in the box for both pictures that comprise the composite image 12. Highlight the phase contrast image 13. Check the blend with composite image box 14. Click on switch view drop down menu and select composite image view 15. Adjust weighting of each picture in the composite 16. Click OK when the desired picture is obtained Save the image of the composite picture on the desktop to later e-mail. IV. Questions and Discussion: How many live and dead cells were found in the live-dead assay section? Differentiated cells typically decrease in size, and obtain a more spherical shape. What is the trend in area and maximum diameter seen between differentiated and undifferentiated cells? Why did you choose pixel as the basic subunit of measurement? What defines the specific size of an individual pixel? How does creating a composite image help in your understanding of the cytoskeletal system in a cell? What is the principle behind fluorescence microscopy? Turn in all printed sheets and pictures. References: Lecture Notes by Professor P. Moghe Schloss, R.S., Vitolo, J.L., Moghe, P.V. (1999). Flow-mediated cell stress induction in adherent leukocytes is accompanied by modulation of morphology and phagocytic function. Medical and Biological Engineering and Computation; 37: 257-263.