Electronic structure of polarization dependent variously
advertisement

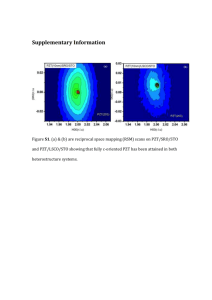
Supplementary materials The crystallographic differences between Pb(Zr1-xTix)O3 (PZT) thin films are attributable to different internal stresses in the films, which may be generated not only by thermal expansion and lattice mismatch between PZT and substrate. The thermal stress is caused by different thermal expansion coefficients between the PZT film and substrate. An internal stress developed when the temperature of the PZT film was reduced from the deposition temperature of about ~800 °C down to the room temperature. The thermal expansion coefficients of PZT and SrTiO3 (STO) were reported to be almost the same,1 indicating that thermal expansion mismatch was not the cause of the internal stress in PZT deposited on STO. In the tetragonal phase, both c-axis elongation and a-axis contraction take place below the Curie temperature (Tc), so that a large tensile strain developed in the c-axis oriented PZT films deposited on STO, which leads to the increase of the c/a ratio. In contrast, Es-Souni et al.2 reported that thermal expansion mismatch was the cause of the internal stress which affects polarization in PZT films grown on silicon and platinized-silicon substrates. In these PZT films, the in-plane tensile stress causes slight differences in a and c lattice constants of the PZT films2,3. References: 1 S. P. Alpay, V. Nagarajan, L. A. Bendersky, M. D. Vaudin, S. Aggarwal, R.Ramesh, and A. L. Roytburd, J. Appl. Phys. 85, 3271 (1999). 2 M. Es-Souni, M. Kuhnke, S. Iakovlev, C.-H. Solterbeck, and A. Piorra, Appl. Phys. Lett. 86, 022907 (2005). 3 W. C. Goh, K. Yao, and C. K. Ong, Appl. Phys. Lett. 87, 072906 (2005). Figure-S1 shows the Fourier transform (FT) of the Ti K-edge extended x-ray absorption fine structure (EXAFS) spectra and the inset shows the FT-amplitude of EXAFS k3χ plot of all PZTs and STO and powder-PbTiO3 (PTO) references. The general line-shapes and radial distributions of the FT spectra of PZTs are not identical to those of the STO and PTO references except <001> PZT, suggesting that the local atomic environments of the Ti atoms in <101> and <111> PZTs are not similar to those of STO and PTO. As for the <001> PZT film, the similarity between its spectrum and that of STO at =0o may be due to that both the <001> PZT film and the STO single crystal have the same tetragonal phase. The first main feature of the FT spectra, corresponding to the distance of nearest-neighbor (NN) ions from the Ti atom, is the fingerprint of 1 the mean inter-atomic Ti-O distances in the TiO6 octahedron. The NN Ti-O inter-atomic distances vary among the three phases in both E//ab and E//c cases. While the <001> tetragonal phase has in-plane and outof-plane NN Ti-O inter-atomic distances of ~1.53 and 1.44 Ǻ, respectively, the <111> rhombohedral phase has corresponding distances of ~1.44 and 1.63 Ǻ. The in- and out-of-plane inter-atomic distances are ~1.50 and 1.60 Ǻ, respectively, for the <101> monoclinic phases, which are intermediate between those of the <001> and <111> phases. Noticed that the first main feature occurs at 1.4-1.6 Ǻ, while the NN Ti-O bond lengths in PZTs is more like 1.8-2.2 Ǻ. This is not an error, but is due to no including the scattering phaseshift when transforming EXAFS data to the FT spectrum, this phase-shift is typically 0.5Ǻ or so. The second main feature at ~3.25Ǻ is observed only in the tetragonal <001> orientated PZT and is similar to that in reference STO (~3.10Ǻ). Thus, the second main feature can be due to the STO substrate, because the La0.67Sr0..33MnO3 (LSMO) buffer layer doesn’t have Ti ions to contribute to the Ti K-edge EXAFS spectrum and the single crystal STO <100>-substrate is the dominant component of the <001> PZT sample. Camargo et al.4 observed a similar second feature in the tetragonal phase of PTO and PZT powders, suggesting that the second feature in <001> PZT may be also related to the tetragonal phase of PZT. On a microscopic scale, the ferroelectric property or polarization is related to collective off-center distortion in transition metal octahedrons. In PZTs, the Ti atom is surrounded by six O atoms to form an octahedron. Without off-center distortion, the Ti ion is located at the center of the six surrounding O ions, which doesn’t form a local electric dipole and there is no polarization or ferroelectricity. When the positively charged Ti ion moves out of the center of the six surrounding negatively charged O ions, a local electric dipole is formed. If all Ti and Zr octahedrons collectively have the same direction of off-center movement and the resultant attractive dipole-dipole interaction energy outweighs the positive elastic energy due to distortion, a spontaneous polarization will be formed and the material is ferroelectric.5 EXAFS measurements show that for the <001>-oriented PZT film, the out-of-plane Ti-O bond distance is shortened, which seems to suggest that the off-center movement of the Ti ion is dominantly along the c-axis, say towards the O ion at the top apex of the octahedron. If this was the case and the octahedron was not distorted, the distance between the Ti 2 ion and the O ion at the bottom apex of the octahedron will be elongated. However, only two bond lengths were observed, i.e. one shortened and another un-shortened with a larger intensity. In ternary semiconductor alloys, say Zn1-xCdxSe, the Zn-Se and Cd-Se bond lengths were shown to approximately retain the values of their respective binary semiconductors ZnSe and CdSe.6 To do so, the tetrahedron in Zn1-xCdxSe, has to be distorted. This property stems from the fact that the distortion of the bond angle costs much less in energy than the alteration of the bond length. With this property in mind one may argue by analogy that the octahedron in PZT has a similar property, i.e. although one of the two Ti-O bonds along the c-axis is slightly shortened due to spontaneous polarization, another Ti-O bond still retains its original bond length without elongation, because elongation costs more energy than distortion of the octahedron. Thus, the distance between the Ti ion and the O ion at the bottom apex of the octahedron will concise with those of in-ab-plane Ti-O bonds, so that only two Ti-O bond lengths were observed. For both <101>- and <111>-oriented PZT films, the EXAFS results show that the in-plane Ti-O bond lengths are slightly shortened. Based on the above argument, one would say that these two PZT films are dominantly polarized in the ab-plane. However, in general for ferroelectric materials with perovskite-like structures, the polarization is along the <111> direction. Thus, the directions of spontaneous polarizations as inferred from the shortening of out-of- or inplane Ti-O bonds may not be conclusive. The deviation between in-plane and out-of-plan Ti-O bond distances may be a measure of the magnitude of the off-center displacement of the Ti ion. 3 Ti K-edge k3 Detector 10 α+θ=90o α θ 0 -10 E 4 PZT 6 8 10 k(Å-1) Magnitude of Transforms = 0 = 70 STO PZT<001> PZT<101> PZT<111> PTO 0 2 4 Radial Coordinate (Å) 6 Fig. S1 FT amplitudes of Ti K-edge EXAFS k3χ from k= 3 to 10 Å-1 at incident angles = 0o and 70o for the three PZTs. The data for reference STO and PTO (powder) were obtained at an angle of incidence of 45o. The right inset shows corresponding oscillations. The left inset presents the experimental geometry. References: 4 E. R. Camargo, E. Longo, E. R. Leite, and V. R. Mastelaro, J. Soid State Chem. 177, 1994 (2004). 5 M.-H. Tsai, Y.-H. Tang, and S. K. Dey, J. Phys.: Condens. Matter 15, 7901 (2003) 6 M. -H. Tsai, S. Lee, F. C. Peiris, and J. K. Furdyna, Phys. Rev. B 65, 235202 (2002) 4 Table I Calculated Born effective charges of the cations in PZT (PbZr0.5Ti0.5O3) in three different crystal structures. Phase Tetragonal Monoclinic Rhombohedral a Cations Z*xx Z*yy Z*zz Pb 3.91 (3.91a) 3.91 (3.91a) 3.86 (3.87a) Ti 6.72 (6.51a) 6.72 (6.51a) 6.65 (6.36a) Zr 6.44 (6.33a) 6.44 (6.33a) 6.50 (6.40a) Pb 4.28 3.92 3.61 Ti 6.76 7.09 6.72 Zr 6.56 6.37 6.57 Pb 3.92 3.92 3.92 Ti 6.70 6.70 6.70 Zr 6.63 6.63 6.63 Reference [22] 5