ASO IgH RQ PCR
advertisement

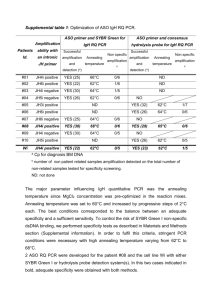
Supplementary information: Materials and methods Samples and nucleic acid extraction Bone marrow (BM) and peripheral blood (PB) samples were obtained from 10 multiple myeloma (MM) patients during standard diagnosis procedure. A lymphoblastoid cell line CL-Wi-L2-729-HF2 1 was used to validate the quantitative PCR methodologies. Samples from healthy PB volunteers were used as negative controls for ASO PCR. An additional specificity control was performed using different pathological cellular samples (BM, PB, leukaphereses, lymphnodes) non-related to the patients for whom an ASO PCR was developed. PB and BM mononuclear cells (MNC) were separated by density gradient centrifugation (Lymphoprep, AbCys, Paris, France, density = 1.077). DNA was isolated using Qiamp mini kit (Qiagen, Courtaboeuf, France) according to the manufacturer’s recommendations. DNA concentration was established with a spectrophotometer. Tumor IgH rearrangement characterization by GeneScanning A single round IgH consensus PCR was performed with 6 VH-family specific primers used in combination with JH fluorescent primers under previously described conditions 2 (Table 1). PCR products were separated via highly sensitive capillary electrophoresis on an automated sequencer (Applied Biosystems, ABI 310, Courtaboeuf, France) and analysed by GeneScan software v. 3.1.2 (ABI). This procedure allowed the detection of MM monoclonal rearrangements. Table 1: PCR primers and hydrolysis probes Primers and probes location Albumine IgH FR1 IgH FR4 Primers designation IgH FR4 probes Primers lenght Reference ALB+ ALB- CTg AgA Agg AgA gAC AAA TC TgC TAT CAA gAC TAC AAA CAC 20 bp 21 bp in-house design VH1 VH2 CCT CAg TgA Agg TCT CCT gCA Agg gAg TCT ggT CCT gCg CTg gTg AAA 24 bp 24 bp 3 VH3 ggT CCC TgA gAC TCT CCT gTg CA 23 bp 3 VH4 TCg gAg ACC CTg TCC CTC ACC TgC 24 bp 3 VH5 gAA AAA gCC Cgg ggA gTC TCT gAA 24 bp 3 VH6 gAA AAA gCC Cgg ggA gTC TCT gAA 24 bp 3 JHD JHEXT ACC TgA ggA gAC ggT gAC CAg ggT g TgA ggA CTC ACC TgA ggA g 20 bp 20 bp 3 JH6 ACC TgA ggA gAC ggT gAC CgT ggT 24 bp 6 JH3i Agg CAg AAg gAA AgC CAT CTT AC 23 bp 7 CAg AgT TAA AgC Agg AgA gag gTT gT 26 bp 7 JH5i AgA gAg ggg gTg gTg Agg ACT 21 bp 7 JH6i gCA gAA AAC AAA GGC CCT AgA gT 24 bp 7 JH1,2,4,5 JH3 CCC Tgg TCA CCg TCT CCT CAg gTg 24 bp CAA ggg ACA ATg gTC ACC gTC TCT TCA 27 bp 7 JH6 CAC ggT CAC CgT CTC CTC Agg TAA Gaa 27 bp 7 IgH intronic JH4i JH Sequence 5'-3' 4 5 7 ASO primer design Monoclonal purified IgH consensus PCR products were directly sequenced with the Big Dye terminator reaction kit (Applied Biosystems) and analysed on an ABI 310. IgH tumor rearrangement sequence was compared to the germline sequence from Ig specific data bases (VBASE : http : //www.mrc.cpe.cam.ac.uk 8 and IMGT : http : //www. imgt.cines.fr 9). The ASO primer was derived from the most hypervariable region CDR3 as shown on figure 1. Figure 1 : IgH CDR3 junctional sequence. VH N V5-51 #01 TGTGCGAGAC TGTGCGAAAG AGGATACAGCATTACT TGTGCGAGACA TATTTCTCTGCCTTATCGGGGGA ACGATATTTTGACTGG TGTGCGAGGGA V3-74 #06 TATTACTGCGCAAG TGTGCGAGC GAGAGAGTGGGACCTAC ATTACTGTGCGAAA V3-23 #10 TGTGCGCGAG TGTGCGAGAG ACCACCTGCACAATAAC CTTTGAAGACTGG JH5 GG D2-21 CCT CTTTGACTACTGG JH4 GGG D2-08 (inv) CTGGTAAATGTGATGTG V1-02 WI AGATTGTATTAATGGTGTATGCTATAC CTTACTACTACTACT JH4 AAGGGGGG D2-08 ACGGTCCGGGGGTC ACTACCACTATGG JH6 GA D1-26 TGTACTAGAG V3-30 #09 GGATTCAAGTTTG TGATATCTGG JH6 TAGAATCTCTAA D3-16 or D5-5 GG V3-49 #08 GCGTTACGATTTGTGGAATGGTTATT TGACCACTGG JH3 TCTCCCGGC D3-03 TAGGGG V4-04 #07 TAACAAGTCGTCC TACCTTGACTCCTGG JH4 CGC D6-06 CCTGTATGAGG TTCGGTATGGACGTCTG JH4 CTGGGGGGGA D3-9 V3-33 #05 TTTGACTCTTGG JH6 GCTG D2-08 (inv) GGATTGTCCCGGGGG JH JH4 A D3-16 V4-39 #04 GTATAACGATCTTTTGGCGGG GGAATGATGC TGTGTCAGAG V3-23 #03 N D3-09 CTAA V3-13 #02 D CATATTGTGGTTATGACTGCTATT GATTCGACCCCTGG JH4 ACTTCTTTGACTACTGG VH, D and JH genes were determined after alignment with germline sequences from databanks. All IgH tumor rearrangement sequences were in frame and submitted to the process of somatic mutations. For the 10 patient rearrangements, a D segment was identified presenting less than 4 mismatches with the germline D sequence. The length of the N nucleotides insertion varied from 0 to 23 nucleotides. The ASO primer (represented in bold and underlined by an arrow in the figure) was designed with OLIGO 3.4 software (W. Richlik, Molecular Biology Insights, Inc., Cascade, CO, USA). The ASO primer choice was guided following definite criteria : first, it should overlap the V-D or D-J junction; second, the Tm should be equivalent to that of the consensus primer; third, the 3’end should be located in a N or D gene segment. Oligonucleotides likely to form secondary structure were avoided. In most cases, these criteria restricted the ASO primer location possibilities. Primers for IgH ASO PCR were chosen either with an ASO downstream or upstream oriented primer. For the CL-Wi-L2-729-HF2 cell line, both primer orientations were assessed. Primers were purchased by Proligo (Paris, France) ASO IgH RQ PCR Two different detection methods were attempted, either with SYBR Green I or with hydrolysis probes (when applicable). All PCR reactions were run in duplicate glass capillaries on the Lightcycler (Roche Applied Science, Meylan, France). The data were analysed by the quantification software (version 3.3, Roche Applied Science). ASO IgH SYBR Green PCR The PCR reactions were performed in 20 L total volume, with « Quantitect SYBR Green I » kit (Qiagen, Courtaboeuf, France) including all PCR reagents with a pre-optimized MgCl2 concentration, 10 pmoles of each primer and 200 ng DNA. The thermal cycling conditions included 15 minutes at 95°C, followed by 45 cycles of 95°C for 15 seconds, optimized annealing temperature for 30 seconds and 72°C for 30 seconds. The fluorescence signal was measured at the end of each elongation step. The final PCR cycle was followed by a melting curve analysis to confirm PCR product identity and to differentiate it from non-specific products. ASO primer and consensus hydrolysis probe for IgH PCR PCR reaction was performed between ASO primer and one of the JH specific intronic primers with 3 different probes which hybridized to the FR4 region, as described by Verhagen 7 (Table 1). A maximum of 500 ng DNA was amplified with 2X “Quantitect reaction mix”, 8 pmoles of each primer and 4 pmoles of probe in a final volume of 20 µL. The “Quantitect” reaction mix (Qiagen, Courtaboeuf, France) included the “hot start” polymerase, adequate buffer, dNTP and a pre-optimized MgCl2 solution. The thermal cycling conditions included 15 minutes at 95°C followed by 45 cycles of 95°C for 10 seconds and optimized hybridization temperature for 60 seconds. The fluorescence was monitored after the annealing/elongation step. Reference gene normalization All samples were normalized by a reference gene in order to take into account the quality and quantity of the DNA input. The albumin gene was chosen for the SYBR Green strategy and the GAPDH gene for the hydrolysis probe strategy. Albumin gene PCRs were performed with a previously optimized PCR using the « LC Hot Start DNA master SYBR Green I » (Roche Applied Science) and 4 mM MgCl2. The thermal cycling conditions included 10 minutes at 95°C, followed by 45 cycles of 95°C for 10 seconds, 60°C for 10 seconds and 72°C for 5 seconds. At the end, a melting curve was performed. The quantification of the GAPDH reference gene was carried out using the “Quantitect” kit with 8 pmoles primers and 4 pmoles probe from “GAPDH Endogeneous reference kit” (Eurogentec, Seraing, Belgium). PCR conditions were as described for the hydrolysis probe ASO IgH RQ PCR with a 60°C annealing/elongation step. Specificity and efficiency determination The specificity of the generated PCR products was determined using several controls. For each ASO PCR, some amplified products were checked by electrophoresis on GelStar stained 1.5% Agarose gels. GelStar and Nusieve agarose were provided by TEBU (Le Perray-en-Yvelines, France). For all ASO PCRs, the size of ASO LightCycler PCR product, estimated by agarose gel electrophoresis, matched the expected length according to the IgH rearrangement sequence (data not shown) thus ensuring the absence of non-specific amplifications resulting from primer’s binding to other gene locus or from primers dimerization. For evaluation of co-amplification of normal and tumor rearrangements, 2 other tests were used : a specificity control was included in each PCR that consisted of a pooled DNA from 4 healthy PB volunteers. Moreover, a specificity test including cell samples non-related to the patients studied was performed for each optimized ASO PCR. For this test, a minimum of 5 samples from different origins (leukaphereses, lymphnode, BM,…) and from different hematological B-cell pathologies were analysed. Nonspecific amplifications were admitted when the crossing point (Cp) for non-related samples were at least over 3 cycles from the sensitivity limit 10 and when they were not frequent (supplemental table 1). Standard curve was generated in order to determine the PCR efficiency and the sensitivity limit of each ASO PCR (see supplementary table 2). The DNA from BM at diagnosis was 8-fold serially diluted in pooled DNA obtained from 4 normal PB volunteers. A constant total DNA input of 200 ng was used for the dilutions. For reference gene PCR, initial BM DNA dilutions in sterile water were used for the standard curve. Since the accurate proportion of tumor cells is not available in the initial BM sample, this sample taken during diagnosis procedure was considered as the calibrator sample for which the tumor load was set to 1 and all samples were referred to it. Reproducibility of the standard curves for one ASO IgH PCR was tested by running the calibration curve 3 times over a period of 3 months (see supplemental table 3). Relative quantification The relative amount of tumor IgH rearrangement and reference gene were calculated with the comparative Ct method (when applicable) and the relative quantification software “RelQuant” v.1.0. (Roche Applied Science). The comparative Ct (Cycle threshold) method The relative concentration of one target compared to another is reflected by the difference in cycle number necessary to achieve the same level of fluorescence. In the comparative Ct method which has been described by Applied Biosystems User Bulletin No.2 (P/N 4303859), the amount of target, normalized to a reference gene and relative to a calibrator is given by : 2 -Ct (with Ct = Ct calibrator sampleCt unknown sample and Ct = Ct of the target gene – Ct of the reference gene). For this method to be valid, equivalent efficiencies between target gene and reference gene PCRs were presumed. Therefore, a validation experiment was conducted by determining the Ct variation with template dilution (supplemental table 6). RelQuant software The RelQuant software was chosen because it takes the efficiency differences between ASO and reference gene PCRs into account. The efficiencies were determined once by using dilutions of BM DNA at diagnosis for IgH ASO and reference gene PCR. The RelQuant software generates a coefficient file in order to save the standard curve parameters that will be used for the analysis of consecutive clinical samples. For each residual disease sample, results are expressed as the target/reference sample ratio divided by the target/reference calibrator ratio and therefore corrected for PCR variations. Aliquots of the calibrator samples corresponding to the BM samples at presentation were frozen in order to guarantee the stability of the calibrator DNA. Absolute quantification with internal calibration curve The calculation of data is based on Cp values obtained from LightCycler software 3.5 by the second derivative maximum method. PCR for standard curve and unknown samples were performed simultaneously in the same run. The LightCycler program extrapolates sample concentration from the standard curve. All samples were analysed in duplicate. The mean extrapolated concentration and standard deviation were calculated automatically. The ratio between ASO PCR concentration and reference gene concentration was established (supplemental table 5). Flow cytometry analysis 106 BM MNC were incubated with anti CD45 PerCP and anti CD38 FITC antibodies (Becton Dickinson, Le Pont de Claix, France) for 30 min at 4°C. After a red PB cell lysis step, cells were washed twice and 100000 cells were acquired using a FACS Calibur flow cytometer (Becton Dickinson). Adequate controls were included. The tumor plasma cells were defined using a combination of high CD38 and low or intermediate CD45 fluorescence 11. References 1.Heitzmann JG, Cohn M. The WI-L2-729-HF2 human hybridoma system. Stable hybrids at high frequency. Mol Biol Med 1983; 1: 235-243. 2.Welterlin V, Debecker A, Tschieb D, Zanetti C, Lange W and Hénon PR. Improvement of clonality detection rate in multiple myeloma using fluorescent IgH PCR with different sets of primers. J Hematother Stem Cell Res. 2000; 9: 983-991. 3.Deane M, Norton JD. Immunoglobulin gene “fingerprinting”: an approach to analysis of B lymphoid clonality in lymphoproliferative disorders. Br J Haematol. 1991; 77: 274-281. 21 4.Owen RG, Johnson RJ, Rawstron AC, Evans PA, Jack A, Smith GM, child JA, Morgan GJ. Assessment of IgH PCR strategies in multiple myeloma. J Clin Pathol. 1996; 49: 672-675. 22 5.Billadeau D, Blackstadt M, Greipp P, Kyle RA, Oken MM, Kay N, Van Ness B. Analysis of B-lymphoid malignancies using allele-specific polymerase chain reaction: a technique for sequential quantification of residual disease. Blood. 1991; 78: 3021-3029. 23 6.Aubin J, Davi F, Nguyen-Salomon F, Leboeuf D, Debert C, Taher M, Valensi F, Canioni D, Brousse N, Varet B. Description of a novel FR1 IgH PCR strategy and its comparison with three other strategies for the detection of clonality in B cell malignancies. Leukemia. 1995; 9: 471-479. 24 7.Verhagen OJ, Willemse MJ, Breunis WB, Wijkhuis AJ, Jacobs DC, Joosten SA, Van Wering ER, Van Dongen JJ, Van der Schoot CE. Application of germline IGH probes in real-time quantitative PCR for the detection of minimal residual disease in acute lymphoblastic leukemia. Leukemia. 2000; 14: 1426-1435 9 8.Cook GP, Tomlinson IM. The human immunoglobulin VH repertoire. Immunol Today. 1995; 16: 237-242. 9.Lefranc MP. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 2001; 29: 207-209. 10. Van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, Van Dongen JJ. Detection of minimal residual disease in hematologic malignancies by real- time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003; 17: 1013-1034. 11. Witzig TE, Meyers C, Therneau T, Greipp PR. A prospective study of CD38/CD45 flow cytometry and immunofluoresence microscopy to detect blood plasma cells in patients with plasma cell proliferative disorders. Leuk Lymphoma. 2000; 38: 345-350.