1 - Computer Science
advertisement

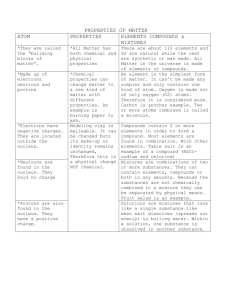
Applications of Machine Learning Approaches Integrating Analytic Methods and Statistics with High Dimensional Visualizations to Different Problems in Cancer Diagnosis and Detection [LIST OFAUTHORS SUBJECT TO DATASETS USED AND WHO WRITES OR HAS DONE ANALYSIS] John McCarthy*, Kenneth A. Marx, Philip O’Neil, M.L. Ujwal, Patrick Hoffman, Alex Gee and Natasha Markuzon AnVil, Inc. 25 Corporate Drive Burlington, MA 01803 *corresponding author jmccarthy@anvilinfo.com; (781) 272-1600 X 460 Abstract Introduction to Data Analysis by Machine Learning Overview of Clustering Methods and Cluster Comparison. Clustering is a method of unsupervised learning. In supervised learning, the object is to learn predetermined class assignments from other data attributes. For example, given a set of gene expression data for samples with known diseases, a supervised learning algorithm might learn to classify disease states based on patterns of gene expression. In unsupervised learning, there either are no predetermined classes or class assignments are ignored. Cluster analysis is the process by which data objects are grouped together based on some relationship defined between objects. It is an attempt to discover novel relationships within a given dataset independent of a priori knowledge about the data space [1,2]. An understanding of relationships between objects is inherent in any clustering technique. This is encoded in a distance measure or distance metric (sometimes called a similarity measure or dissimilarity measure). Unlike a distance measure, a distance metric is required to satisfy the triangle inequality. The most commonly used distance metric is Euclidean distance, which in three dimensions corresponds to the physical distance between objects in space. The Manhattan distance is a “city block” measure, in contrast to the Euclidean “shortest distance between two points.” There are several other alternatives including correlation measures. Generally, each measure defines a relationship based on certain assumptions about the underlying data and attempts to capture particular data characteristics [3]. In conjunction with selecting a distance measure, one must also decide on a clustering algorithm, the procedure by which these n-dimensional objects are grouped together to form clusters. Classical clustering techniques are divided into two groups, the partitioning methods and the hierarchical approaches [1, 3-5]. In more recent years, a third group of techniques that includes probabilistic approaches has emerged [6]. Similar to the choice of distance metric, the selection of clustering algorithms for specific datasets poses problems, as each algorithm focuses on certain types of relationships within any given dataset, some overlapping and others unique. The partitioning methods, sometimes referred to as iterative relocation algorithms, construct clusters by first partitioning the data objects into some number of clusters and then recursively moving objects between clusters until some cluster measure is minimized. The result is a set of object groups where each object is assigned to only one cluster. Hierarchical approaches are based on tree structures where the data objects occupy the leaves of the tree and the nodes of the tree define the relationship between subtrees or leaves. These hierarchical methods are defined as either agglomerative (bottom-up) or divisive (top-down) approaches. Agglomerative techniques start with each object in a separate cluster and perform a series of successive fusions of clusters into larger clusters. Divisive methods start with all objects in a single cluster and provide successive refinements of the clusters into smaller clusters. Agglomerative methods include: nearest neighbor (single-link method), furthest neighbor (complete-linkage method), centroid cluster analysis, median cluster analysis, group average method, Ward’s method, McQuitty’s methods, Lance and Williams flexible method, and others [7]. Divisive methods include monothetic methods, which are based on the value of a single attribute, and polythetic, which are based on the values of all attributes. Hierarchical techniques provide no indicators on the number of clusters that the data should be clustered into. The tree structure can be cut at various levels and the resulting subtrees determine the clusters and their number. Probabilistic techniques provide information as to how well an object belongs to each cluster rather than just providing the cluster memberships. Given that most data spaces do not contain well-defined objects, probabilistic techniques provide additional information about a data space. Examples of probabilistic techniques include the fuzzy clustering algorithms [6]. Many additional clustering techniques are a mixture of the basic types of methods discussed above. Over the past five years there have been a significant number of academic and commercial clustering and classification approaches focused on high dimensional data, particularly biological and chemical data. Fasulo [4] for example, describes recent results on clustering, each of which approaches the clustering problem from a different perspective and with different goals. A fundamental question that arises repeatedly is which clustering technique is better? The answer to this question is an important commercial consideration for pharmaceutical and biotechnology companies when the $ 500-800 million average cost of developing a successful drug is at stake. It is even more important when the outcome is accurate clinical detection of a specific cancer in a patient. In drug discovery, the outcome of a clustering technique can influence decisions about selecting drug targets and chemical lead compounds. Several investigators, including Schaffer [8], Felders [9], Dietterich [10] and Cheng [11], have attempted to answer this question. In our view, the assessment of which clustering technique is better is domain and data dependent, given the incomplete information on which clusters are usually based. The question of which technique is better is not the correct question to ask. A number of recent papers have discussed the pitfalls of current comparative analyses, especially when using public domain datasets and databases [12]. Combining Contextual Knowledge with Experimental Data in the Mining of Microarry Gene Expression and Other Molecular Datasets. The availability of genome-wide expression profiles promises to have a profound impact on the understanding of basic cellular processes, the diagnosis and treatment, and the efficacy of designing and delivering targeted therapeutics. Particularly relevant to these objectives is the ability to cross-reference experimental and analytical results with previously known biological facts, hypotheses, theories and results. Biological and biomedical literature databases provide the kind of knowledge warehouses for such extensive crossreferencing. However, the volume of such databases makes the task of cross-referencing lengthy, tedious and daunting [13]. In order to explain the underlying biological mechanisms and assign “biological meaning” to clusters of genes obtained by analytical methods, it is necessary to crossreference genes with external information sources. Efforts in this direction are particularly relevant as to clustering/classification methods which typically rediscover known associations between genes. It is therefore important to take full advantage of the existing knowledge about classical cellular pathways, including the metabolic and signaling pathways, transcription factors, regulatory elements/motifs in sequence or structure information, and assigned gene functions. Literature databases, which are a rich source of information can be used to discover and analyze biologically significant information based on co-citations or co-occurrences of pairs of genes: gene terms or gene: disease terms in a given scientific paper. Likewise, one can extract biologically meaningful relationships in the semantic framework of ontologies being specifically developed to capture such information that use experimental results reported in literature [14]. One of AnVil’s strengths is our ability to carry out integrated data mining and visualization analyses on large, complex nonlinear datasets that may have as many as 50,000 data dimensions. Therefore, we have a practical way to overcome the need to reduce dimensionality early on in addressing any specific problem. One advantage this mechanism provides is the ability to simultaneously handle large numbers of data dimensions, enabling us, for example, to add contextual knowledge into already largedimensionality datasets that researchers have to analyze; the contextual knowledge is simply considered as additional data dimensions. We discuss the distinct advantages of our technology in greater detail in the following sections. The Importance of High-dimensional Data Visualization and its Integration with Analytic Data Mining Techniques. Visualization, data mining, statistics, as well as mathematical modeling and simulation are all methodologies that can be used to enhance the discovery process [15]. AnVil’s expertise lies in a combination of analytic data mining techniques integrated with advanced high-dimensional visualizations (HDVs). There are numerous visualizations and a good number of valuable taxonomies (See [16] for an overview of taxonomies). Most information visualization systems focus on tables of numerical data (rows and columns), such as 2D and 3D scatterplots [17], although many of the techniques apply to categorical data. Looking at the taxonomies, the following stand out as high-dimensional visualizations: Matrix of scatterplots [17]; Heat maps [17]; Height maps [17]; Table lens [18]; Survey plots [19]; Iconographic displays [20]; Dimensional stacking (general logic diagrams) [21]; parallel coordinates [22]; Pixel techniques, circle segments [23]; Multidimensional scaling [23]; Sammon plots [24]; Polar charts [17]; RadViz [25]; Principal component analysis [26]; Principal curve analysis [27]; Grand Tours [28]; Projection pursuit [29]; Kohonen self-organizing maps [30]. Grinstein et.al., [31] have compared the capabilities of most of these visualizations. Historically, static displays include histograms, scatterplots, and large numbers of their extensions. These can be seen in most commercial graphics and statistical packages (Spotfire, S-PLUS, SPSS, SAS, MATLAB, Clementine, Partek, Visual Insight’s Advisor, and SGI’s Mineset, to name a few). Most software packages provide limited features that allow interactive and dynamic querying of data. HDVs have been limited to research applications and have not been incorporated into many commercial products. However, HDVs are extremely useful because they provide insight during the analysis process and guide the user to more targeted queries. Visualizations fall into two main categories: (1) low-dimensional, which includes scatterplots, with from 2-9 variables (fields, columns, parameters) and (2) highdimensional, with 100-1000+ variables. Parallel coordinates or a spider chart or radar display in Microsoft Excel can display up to 100 dimensions, but place a limit on the number of records that can be interpreted. There are a few visualizations that deal with a large number (>100) of dimensions quite well: Heatmaps, Heightmaps, Iconographic Displays, Pixel Displays, Parallel Coordinates, Survey Plots, and RadViz. When more than 1000 records are displayed, the lines overlap and cannot be distinguished. Of these, only RadViz is uniquely capable of dealing with ultra–high-dimensional (>10,000 dimensions) datasets, and we discuss it in detail below. RadViz™ is a visualization and classification tool that uses a spring analogy for placement of data points and incorporates machine learning feature reduction techniques as selectable algorithms. 13-15 The “force” that any feature exerts on a sample point is determined by Hooke’s law: f kd . The spring constant, k, ranging from 0.0 to1.0 is the value of the feature for that sample, and d is the distance between the sample point and the perimeter point on the RadViz circle assigned to that feature-see Figure 1. The placement of a sample point, as described in Figure 1 is determined by the point where the total force determined vectorially from all features is 0. The RadViz display combines the n data dimensions into a single point for the purpose of clustering, but it also integrates analytic embedded algorithms in order to intelligently select and radially arrange the dimensional axes. This arrangement is performed through Autolayout, a unique, proprietary set of algorithmic features based upon the dimensions’ significance statistics that optimizes clustering by optimizing the distance separating clusters of points. The default arrangement is to have all features equally spaced around the perimeter of the circle, but the feature reduction and class discrimination algorithms arrange the features unevenly in order to increase the separation of different classes of sample points. The feature reduction technique used in all figures in the present work is based on the t statistic with Bonferroni correction for multiple tests. The circle is divided into n equal sectors or “pie slices,” one for each class. Features assigned to each class are spaced evenly within the sector for that class, counterclockwise in order of significance (as determined by the t statistic, comparing samples in the class with all other samples). As an example, for a 3 class problem, features are assigned to class 1 based on the sample’s t-statistic, comparing class 1 samples with class 2 and 3 samples combined. Class 2 features are assigned based on the t-statistic comparing class 2 values with class 1 and 3 combined values, and Class 3 features are assigned based on the t-statistic comparing class 3 values with class 1 and class 2 combined. Occasionally, when large portions of the perimeter of the circle have no features assigned to them, the data points would all cluster on one side of the circle, pulled by the unbalanced force of the features present in other sectors. In this case, a variation of the spring force calculation is used, where the features present are effectively divided into qualitatively different forces comprised of high and low k value classes. This is done via requiring k to range from – 1.0 to 1.0. The net effect is to make some of the features pull (high or +k values) and others ‘push’ (low or –k values) the points to spread them absolutely into the display space, but maintaining the relative point separations.It should be stated that one can simply do feature reduction by choosing the top features by t-statistic significance and then apply those features to a standard classification algorithm. The t-statistic significance is a standard method for feature reduction in machine learning approaches, independently of RadViz. The top significance chemicals selected with the t-statistic are the same as those selected by RadViz. RadViz has this machine learning feature embedded in it and is responsible for the selections carried out here. The advantage of RadViz is that one immediately sees a “visual” clustering of the results of the t-statistic selection. Generally, the amount of visual class separation correlates to the accuracy of any classifier built from the reduced features. The additional advantage to this visualization is that sub clusters, outliers and misclassified points can quickly be seen in the graphical layout. One of the standard techniques to visualize clusters or class labels is to perform a Principle Component Analysis and show the points in a 2d or 3d scatter plot using the first few Principle Components as axes. Often this display shows clear class separation, but the most important features contributing to the PCA are not easily seen. RadViz is a “visual” classifier that can help one understand important features and how many features are related. And RadViz Figure 1. How it works We have studied the following systems related to cancer detection: 1. GI50 compound 60 cancer cell lines 2. Microarray lung cancer data 3. proteomics MS dataset 4. UNOS 1. Data Mining the NCI Cancer Cell Line Compound GI50 Data Set using Supervised Learning Techniques Introduction. In a data mining study of 8 large chemical structure databases, it was observed that the NCI Developmental Therapeutics Programs’data set contained by far the largest number of unique compounds of all the databases (32). The NCI compound data set has been mined in a series of reports by the intramural NCI Informatics research group of Weinstein and collaborators. Supervised learning via cluster correlation, principle component analysis and various neural network techniques have all been applied, as well as statistical techniques (33,34). Many literature citations have described compound class subsets, such as: tubulin active compounds (35), pyrimidine biosynthesis inhibitors (36) and topoisomerase II inhibitors (37), that possess similar mechanisms of action (MOA), share similar structures or develop similar patterns of drug resistance. Compound structure classes such as the ellipticine derivatives have also been studied and point to the validity of the concept that fingerprint patterns of activity in the NCI data set encode information concerning MOAs and other biological behavior of tested compounds (38). More recently, gene expression analysis has been added to the data mining activity of the NCI compound data set (39). Gene expression profiles of the 60 cancer cell lines have been employed in a method that predicted chemosensitivity, using the GI50 value data, for a few hundred compound subset of the NCI data set (40). After we completed our analysis (41), gene expression data on the 60 cancer cell lines was combined with NCI compound GI50 data and with a 27,000 feature database computed for the NCI compounds to calculate chemical features similar to those identified in the following study and as we have presented elsewhere (42). Here we use microarray based gene expression data to first establish a number of ‘functional’ classes of the 60 cancer cell lines. These functional classes are then used in a series of 2-Class supervised learning problems, using a subset of 1400 of the NCI compounds’ GI50 values as the input to a clustering algorithm in the RadViz™ program (43). At p < .01 significance, RadViz™ identifies two small compound subsets that accurately classify the cancer cell line classes: melanoma from non-melanoma and leukemia from non-leukemia, as we have previously reported (41). We then demonstrate that independent analytic classifiers validate the two small compound subsets we selected. We found them to both be significantly enriched in quinone compounds of two distinct subtypes that we relate to the literature. Specific Methods Used. For the ~ 4% missing values found in the 1400 compound data set, we tried and compared two approaches to missing value replacement: 1) record average replacement; 2) multiple imputation using Schafer’s NORM software (44). Using either missing value replacement method for the starting data set, there was close agreement( always > 90%) between the NCI compound lists selected in identical 2-Class Problem classifications we present below. Therefore, in the present study, we used the record average replacement method for all the data presented. Clustering of cell lines was done with R-Project software using the hierarchical clustering algorithm with “average” linkage method and a dissimilarity matrix computed as 1 – the Pearson correlations of the gene expression data. AnVil Corporation’s RadViz™ software (45) was used for feature reduction and initial classification of the cell lines based on the compound GI50 data. The selected features were validated using several classifiers from Weka 3.1.9 (Waikato Environment for Knowledge Analysis, University of Waikato, New Zealand). The classifiers used were IB1 (nearest neighbor), IB3 (3 nearest neighbor), logistic regression, Naïve Bayes Classifier, support vector machine, and neural network with back propagation. Both ChemOffice 6.0 (CambridgeSoft Corp.) and the NCI website were used to identify compound structures via their NSC numbers and substructure searching to identify quinone compounds in the larger data set were carried out using ChemFinder (CambridgeSoft). Results and Discussion Identifying functional cancer cell line classes using gene expression data. Based upon gene expression data, we initially decided to identify cancer cell line classes that we could use in a subsequent supervised learning approach to stringently select compound subsets capable of classifying the individual cancer cell classes. In Figure 2, we present a hierarchical clustering dendrogram using the 1-Pearson distances calculated from the T-Matrix, comprised of 1376 gene expression values determined for the 60 NCI cancer cell lines (43). There are five well defined clusters observed. Four of the clusters in Figure 2 (renal, leukemia, ovarian and colorectal from second left to right) represent pure cell line classes. In only the melanoma class instance does the class contain two members of another clinical tumor type, two breast cancer cell lines - MDA-MB-435 and MDA-N. The 2 breast cancer cell lines behave functionally as melanoma cells and seem to be related to melanoma cell lines via a neuroendocrine origin as has already been observed and remarked upon (43). The remaining cell lines in the Figure 2 dendrogram, those not found in any of the five functional classes, are defined as being in the sixth class- the non- melanoma, leukemia, renal, ovarian, colorectal class. In the supervised learning studies that follow, we treat these six functional clusters as the ground truth. 2-Class Cancer Cell Classifications and Validation of Results. High class number classification problems are difficult to implement where the data are not clearly separable into distinct classes and we could not successfully carry out a 6-class classification of the cancer cell line classes based upon the starting GI50 compound data. For this reason, we chose to implement 3-Class and 2-Class problems utilizing RadViz™ , which combines an analytic class discrimination layout algorithm, employing feature reduction, with a high dimensional visualization resulting from the algorithm’s output (25, 45-47). Starting with the small 1400 compounds’ GI50 data set that contained no missing values for all 60 cell lines, those compounds were selected that were effective in carrying out the classification at the p < .01 (Bonferroni corrected t statistic) significance level. The 3 -Class problem at p < .01 significance, for the melanoma, leukemia and nonmelanoma, non-leukemia classes are presented in Figure 3. In contrast to the 6-Class problem results we obtained (data not shown), the 3-Class problem result in Figure 3 at the same significance, p < .01, produced clear and accurate class separations of the 60 cancer cell lines. There were 14 compounds selected in the 2-Class problem as being most effective (lowest GI50 values) against melanoma class cells and 30 compounds were identified as most effective against the leukemia class cells. Similar classification results were obtained for the separate 2-Class problems melanoma vs. non-melanoma and leukemia vs. non-leukemia (data not shown; [41]). For all other possible 2-Class problems, we found that few to no compounds could be selected at p <.01. Validating the results we obtained from our RadViz™ methodology for compound selection in both 2-Class and the 3-Class problems was our next goal. We utilized 6 analytic classification techniques ( Instance Based 1, Instance based 3, neural networks, logistic regression and support vector machines), with the same selected compounds’ GI50 values as a classifier set based upon calculating the frequency of correct classification and using a 60-fold repetition of the training-test process using the holdone-out method. Well above 90% accuracies were achieved using these compound subsets (data not shown; see [41]). For the 80 compounds selected as most effective against leukemia at the p < .01 criterion, the average accuracy achieved by the 6 analytic algorithms was 99.3%, corresponding to only a 0.7% error rate. Based upon repetitively selecting 80 compounds randomly, the average level of accuracy was calculated to be 95.7%, corresponding to a 4.3% error rate (data not shown). It is counterintuitive to achieve an accuracy as high as 95.7% from any 80 compounds randomly selected from the 1400. However, this is because any 80 randomly selected compounds will always include a small number of the significant compounds. Therefore, using the RadViz™ selected compounds represented a greater than 6-fold lowered level of error compared to the randomly selected compounds, thus validating our selection methodology. Quinone Compound Subtypes preferentially effective against melanoma. Next, we decided to examine the chemical identity of the compounds selected as most effective against melanoma and leukemia. To summarize, for the 14 compounds selected in Figure 3 as most effective against melanoma, 11 are p-quinones. Of the 11 p-quinones, all 11 are internal ring quinone structures. We display a representative example of these structures in Figure 4. These internal ring quinones possess either 2 neighbor aromatic 5 or 6 member fused rings, some of which are heteroatom containing, on either side of the quinone ring or an aromatic fused ring neighbor on one side and covalent non-H substitutions off the other side of the quinone. These substitutions all have electronegative atoms covalently bonded to either or both the o and m positions of the quinone ring, except for one compound which has an –OH substituent off the adjacent ring. In 8 of the cases, the internal ring quinones are directly bonded to 2 electronegative atoms, either heteroatoms contained within the aromatic fused ring or as small covalent substituents. And in 2 more cases, the internal quinone ring is bonded to 1 electronegative atom within a neighboring fused ring. A recent analysis by Blower et.al., (42) simultaneously correlating gene expression data for the 60 cancer cell lines with GI50 values for a set of 4463 compounds for which a 27,000 feature set had been calculated. The investigators identified a subclass of compounds containing a benzothiophenedione core structure that were most highly correlated with the expression patterns of Rab7 and other melanoma specific genes. There is clearly some overlap between the internal quinone subtype we have defined in the present study and the benzothiophenedione core structure members. Out of the 11 internal quinone compounds we identified, 3 are of the benzothiophenedione core structure class, but they are not amongst the most effective compounds we identified. The Rab7 gene is a member of the GTP binding protein family involved in the docking of cellular transport vesicles and is a key regulator of aggregation and fusion of late endocytic lysosomes (48). In the same study, a number of other genes whose expression levels highly correlate with the same compounds, expressed proteins involved in other lysosomal functions, suggesting a link between the quinone oxidation potential, the proton pump and the electron transport chain. These investigators suggested the possibility that benzodithiophenedione compounds may act directly as surrogate oxidizing agents, effectively competing with ubiquinone in the electron transport chain and thereby disrupting an essential cellular redox process. The effectiveness of any compound in this type of mechanism would be based upon its redox potential. Quinone Compound Subtypes preferentially effective against leukemia. There were 30 compounds selected as most effective against leukemia in the leukemia, nonleukemia 2-Class Problem, of which 8 are structures containing p-quinones. In contrast to the internal ring quinones comprising the melanoma class, 6 out of the 8 leukemia pquinones were external ring quinones. We display an example of these structures in Figure 4B. In contrast to the internal ring quinones, these external ring quinones had only one aromatic fused ring neighbor which had no ring heteroatoms in all cases. Also different, the quinone was itself at the periphery of the molecule and had no non-H substituents off the exterior side of the ring at either o or m positions. Thus, the ‘external’ and ‘internal’ quinone rings should possess different electron densities and redox potentials for the quinoid oxygens. Besides redox potentials, other possible subtype differences may exist such as: solubility, steric differences relative to metabolic enzyme active sites, differential cellular adsorption, etc. Again, the recent analysis by Blower, et.al., (42) discussed above, identified a sub-class of compounds, comprised of an indolonaphthoquinone core structure. These compounds were most highly correlated with the expression patterns of LCP1, lymphocyte cytosolic protein 1 (L-plastin located on chromosome 13), HS1, a hematopoietic lineage specific gene, and other leukemia specific genes. There is overlap between the external quinone subtype and the indolonaphthoquinone core structure members. This overlap between the two studies is somewhat remarkable since we included no gene expression data in our analysis of the GI50 values, as did the Blower study (42). This suggests two things. The first is that there is sufficient information inherent in the compound GI50 values to carry out the basic core discovery presented here, without the need to include gene expression data in the analysis. The second is that the class discrimination layout algorithm of RadViz™, used here to select and array compound’s axes to maximize the cluster separation, is a highly effective data mining tool. Uniqueness of Two Quinone Subtypes. In order to ascertain the uniqueness of the two quinone subsets found effective against melanoma and leukemia, we first determined the extent of occurrence of p-quinones of all types in our starting data set of 1400 compounds. To do this, we examined the entire data set via substructure searching using the ChemFinder 6.0 software. We found that the internal and external quinone subtypes we identified as effective against melanoma and leukemia respectively, represent a significant fraction, 25 % (10/41) of all the internal quinones and 40 % (6/15) of all the external quinones in the data set. In addition, we determined that only one compound, NSC 621179, which is not a quinone but an epoxide, was found to be effective against both melanoma and leukemia in a 2-Class classification where one class was both leukemia and melanoma cell lines and the second class was non-melanoma, non-leukemia cell lines. This result attests to the uniqueness of the specificity of the two quinone subtype classes. The NCI data set lists 92 compounds known to fall within one of 6 Mechanism Of Action (MOA) Classes: alkylating agents, antimitotic agents, topoisomerase I inhibitors, topoisomerase II inhibitors, RNA/DNA antimetabolites, DNA antimetabolites (33). We determined that the most effective 14 and 30 compounds against melanoma and leukemia respectively identified in the 2-Class problems do not fall into clusters with any one of these 6 MOA compound classes. Using the 14 melanoma and the 30 leukemia compounds as the two classes, the RadViz™ class discrimination algorithm layed out the 60 cell lines based upon the 2 class compounds’ GI50 values (figure not shown: [42]), producing 2 well separated compound classes. Then the 92 known MOA compounds were simply placed upon this optimized RadViz™ display. None of the 92 compounds in the 6 MOA classes clustered with any of the compounds in either the melanoma or the leukemia class. Sub-classification of Leukemia Cell Lines. We next asked the question whether we could sub-classify either the melanoma or the leukemia cell lines into distinct clinical sub-classes based upon using our 2 respective compound classes. Therefore, we first carried out an unsuccessful 3-Class RadViz™ based classification for the melanotic melanoma, other melanoma and non-melanoma cell classes at p < .05, using the most effective 14 compounds identified as most effective against all melanomas (data not shown).We next carried out a 3-Class RadViz™ based leukemia cell sub-classification for the acute lymphoblastic leukemia (ALL), non-ALL leukemia (other) and nonleukemia cell classes at p < .05. To carry out the sub-classification, we used the most effective 30 compounds identified for the p < .01 selection criterion as most effective against all leukemias and this result is presented in Figure 5. Six of the 30 compounds were most effective against the ALL class; while 12 of the 30 compounds were most effective against the non-ALL leukemia. In this result, it is clear that there is a separation of the 2 ALL cell lines (CCRF-CEM and MOLT-4) from the non-ALL leukemia subclass. These two ALL cell lines were also the most closely clustered leukemia cells in the Figure 2 gene expression based clustering dendrogram. This suggests the interesting possibility that the chemical identity of the compounds most effective against the 2 ALL cell lines are linked to the gene functions most responsible for closely clustering these 2 ALL cell lines in Figure 1. NAD(P)H: quinone oxidoreductase 1 -Quinone substrates and Leukemias Different redox potentials and enzymatic reactivities are likely to be the key to how these quinone subtypes differentially affect melanoma and leukemia cells. In addition to the gene candidates identified as potentially involved in quinone activity in the Blower, et.al, (42) study, a strong candidate enzyme for this differential reactivity is NAD(P)H:quinone oxidoreductase 1 (QRI, NQO1, also DT-diaphorase; EC 1.6.99.2). This enzyme is expressed in normal cells and at high levels in many types of tumors (49). It catalyzes two electron reduction of a variety of substrates with the most efficient substrates being quinones (50). The enzyme has been crystallized and the X-ray structures of the apoenzyme at 1.7-A resolution and its complex with substrate duroquinone (2,5A) are known (51,52). NAD(P)H:quinone oxidoreductase 1 is a chemoprotective enzyme that protects cells from oxidative challenge. Antitumor quinones, of the type we have identified above in the NCI data set, may be bioactivated by this enzyme to forms that are cytotoxic (50). This catalytic property makes this enzyme an excellent target for enzymedirected drug development (52). Reductive activation is particularly well–suited for treatment of hypoxic tumors, where the bioreduction of the chemical agent to hydroquinone cannot be reversed by endogenous oxygen (53). Interestingly, there are a number of reports that correlate altered forms or alleles of this enzyme with leukemia (54-56). These reports, associating leukemias with particular aspects of NAD(P)H:quinone oxidoreductase 1, suggest the enzyme as likely being a significant factor in why the external quinone subtypes, acting as particularly potent and effective substrates, exhibit their differential selectivity toward leukemias. We believe that only through experiments or calculations to determination the redox potentials of the different quinone compounds, influenced by the type and distribution of substituent groups, will the exact nature of the compound selectivity exhibited by the subtypes in our study be known. 2. Microarrays Analysis of High Throughput Gene Expression Experiments: Effects of Normalization Methods on Gene Expression Analysis Clustering Results. Completion of the Human Genome Project has made possible the study of the gene expression levels of over 30,000 genes [14,15; although a ‘final’ human genome sequence is scheduled for release in Spring, 2003]. Major technological advances have made possible the use of DNA microarrays to speed up this analysis. Even though the first microarray experiment was only published in 1995, by October 2002 a PubMed query of microarray literature yielded more than 2300 hits, indicating explosive growth in the use of this powerful technique. DNA microarrays take advantage of the convergence of a number of technologies and developments including: robotics and miniaturization of features to the micron scale (currently 20-200 um surface feature sizes for spotting/printing and immobilizing sequences for hybridization experiments), DNA amplification by PCR, automated and efficient oligonucleotide synthesis and labeling chemistries, and sophisticated bioinformatics approaches. It is this latter aspect of the development of microarray technology that our Phase II proposal addresses. One significant aspect of analyzing microarray gene expression data is the need for normalization to remove non-biological sources of variation (noise), in order to make meaningful comparisons of data from different microarrays. The noise results from differences in individual chips, labeling chemistry, length of immobilized oligonucleotide sequence, different optical properties of various data scanners and other sources. The importance of understanding and controlling these variables has been underscored by the apparent lack of reproducibility of some published microarray studies. This has led to the establishment of the MIAME publication guidelines that detail the following requirements for describing microarray experiments: 1) experimental design, 2) array design and the name and location of array spots, 3) sample name extraction and labeling, 4) hybridization protocols, 5) image measurement methods, 6) controls used [16-18]. Normalization techniques that have been applied include simple linear scaling, locally linear transformations, and other nonlinear methods. To some extent, the techniques used depend on the type of array being used. In 2 channel arrays, for example cDNA microarrays, the issue is primarily within-chip normalization to correct distortions based on location and signal intensity. Between-chip normalization is less of an issue for these arrays because one channel usually contains a reference tissue that is common to all arrays in the experiment. Between-chip normalization has the potential of introducing more noise than it eliminates. A number of thorough discussions of normalization techniques for cDNA arrays have been presented [19,20]. These normalization approaches include dye swap experiments to correct for differences between the two channels, using the lowess function to correct for global intensity based differences (i.e. across all genes on the chip), and using the lowess function locally to account for spatial and print-tip differences. For the majority of applications, Affymetrix microarrays are in use. For these arrays, between-chip normalization is an important issue, and is closely related to the method of calculation of gene expression value from multiple probes for each gene. Techniques proposed for calculating expression include the original Affymetrix method of average difference between perfect match and mismatch probes, the Model Based Expression Index approach of Li and Wong [21], and the Robust Multichip Average approach of Irizzary et al [22]. Durbin et al [23] have suggested a variance-stabilizing transformation to aid microarray analysis. There is the additional consideration of whether to normalize data based on probe level measurements or expression calculations, and whether to use a baseline array for comparison or to normalize over the complete set of data. Bolstad et al [24] present comparisons of some of these techniques. They recommend probe level and complete data methods in general, and quantile normalization in particular. They also found that the invariant set normalization approach of Schadt et al [25] using a baseline array gives results that are comparable to complete data methods. Our experience has shown that quantile normalization works well even when probe level data are not available. However, quantile normalization makes the implicit assumption that the data on all chips have the same distribution. For some datasets this may not be appropriate. Different normalization and modeling techniques can lead to widely varying judgments and interpretations of differential gene expression. In this Phase II proposal, we aim to investigate the effects of different data normalizations on clustering. We will compare quantile normalization, invariant set normalization, lowess local regression, and simple linear scaling. We will focus primarily on Affymetrix type arrays, but we will ensure that the platform we develop supports the adaptation and application of these techniques to two channel microarrays where appropriate. We will also investigate the effects of different modeling techniques on clusters. The more successful a technique is at removing noise, the more likely it is that the clusters generated will be accurate and will have biological meaning. On the other hand, the quality and stability of clusters could be a useful measure of the appropriateness of the normalization and modeling techniques used. Therefore, a goal of this Phase II proposal is to provide users with decision making tools to decide which normalization approach is optimal or close to optimal for a given microarray dataset. Also, the normalization tools will be integrated with the perturbation algorithm output, discussed below, to determine the stability of clusters from different normalizations. In this way, we can provide users with the identity of those genes that are most stable within clusters, and those that are unstable and jump between clusters as a result of different normalizations. 3. Proteomics 4. UNOS Conclusions Acknowledgements AnVil and the authors gratefully acknowledges support from two SBIR Phase I grants R43 CA94429-01 and R43 CA096179-01 from the National Cancer Institute. Also, support is acknowledged from ………..X Y Z References 1. A. Strehl. Relationship-based Clustering and Cluster Ensembles for Highdimensional Data Mining. Dissertation, The University of Texas at Austin, May, 2002. 2. I. H. Witten and E. Frank. Data Mining: Practical Machine Learning Tools and Techniques with Java Implementations. San Francisco: Morgan Kaufmann, 2000. 3. J. A. Hartigan. Clustering Algorithms. New York: John Wiley & Sons, 1975. D. Fasulo. “An Analysis of Recent Work on Clustering Algorithms.” http://www.cs.washington.edu/homes/dfasulo/clustering.ps, April 26, 1999. 5. C. Fraley and A. E. Raftery “Model-Based Clustering, Discrimination Analysis, and Density Estimation.” Technical Report no. 380, Department of Statistics, University of Washington, Seattle, October, 2000. 6. F. Höppner, F. Klawonn, R. Kruse, and T. Runkler. Fuzzy Cluster Analysis: Methods for Classification, Data Analysis and Image Recognition. Chichester: John Wiley & Sons, 1999.. 7. Everitt, B., Cluster Analysis, Halsted Press, New York (1980). 8. Schaffer, C., Selecting a classification method by cross-validation, Machine Learning, 13:135-143 (1993). 9. Feelders A., Verkooijen W.: Which method learns most from the data? Proc. of 5th International Workshop on Artificial Intelligence and Statistics, January 1995, Fort Lauderdale, Florida, pp. 219-225, (1995). 10. Dietterich, T.G., Approximate statistical tests for comparing supervised classification learning algorithms. Neural Computation, 10(7), 1895-1924. 11. Cheng, J., Greiner, R., Comparing Bayesian network classifiers. In Proceedings of the 15th Conference on Uncertainty in Artificial Intelligence (UAI ’99), 101-107, Morgan Kaufmann Publishers (1999). 12. Salzberg, S. L., On Comparing Classifiers: A Critique of Current Research and Methods, Data Mining and Knowledge Discovery, 1999, 1:1-12, Kluwer Academic Publishers, Boston. 13. Ramaswamy, S., Ross, K.N., Lander, E.S. and Golub, T.R. A molecular signature of metastasis in primary solid tumors. Science, 22, 1-5. 14. Chaussabel., D. and Sher, A. Mining microarray expression data by literature profiling. Genomebiology, 3, 1-16 4. 15. Fayyad, U.M., Piatetsky-Shapiro, G., Smyth, P., Uthurusamy, R. (Eds.) Advances in knowledge discovery and data mining, AAAI/MIT Press, 1996. 16. B. Shneiderman, “The Eyes Have It: A Task by Data Type Taxonomy of Information Visualization,” presented at IEEE Symposium on Visual Languages '96, Boulder, CO, 1996. 17. J. W. Tukey, Exploratory Data Analysis. Reading, MA: Addison-Wesley, MA, 1977. 18. R. Rao and S. K. Card, “The Table Lens: Merging Graphical and Symbolic Representations in an Interactive Focus+Context Visualization for Tabular Information,” presented at ACM CHI '94, Boston, MA, 1994. 19. D. F. Andrews, “Plots of High-Dimensional Data,” Biometrics, vol. 29, pp. 125-136, 1972. 20. H. Chernoff, “The Use of Faces to Represent Points in k-Dimensional Space Graphically,” Journal of the American Statistical Association, vol. 68, pp. 361-368, 1973. 21. J. Beddow, “Shape Coding of Multidimensional Data on a Microcomputer Display,” presented at IEEE Visualization '90, San Francisco, CA, 1990. 22. A. Inselberg, “The Plane with Parallel Coordinates,” Special Issue on Computational Geometry: The Visual Computer, vol. 1, pp. 69-91, 1985. 23. D. A. Keim and H.-P. Kriegel, “VisDB: Database Exploration Using Multidimensional Visualization,” IEEE Computer Graphics and Applications, vol. 14, pp. 40-49, 1994. 24. J. W. J. Sammon, “A Nonlinear Mapping for Data Structure Analysis,” IEEE Transactions on Computers, vol. 18, pp. 401-409, 1969. 25. P. Hoffman and G. Grinstein, “Dimensional Anchors: A Graphic Primitive for Multidimensional Multivariate Information Visualizations,” presented at NPIV '99 (Workshop on New Paradigmsn in Information Visualization and Manipulation), 1999. 26. H. Hotelling, “Analysis of a Complex of Statistical Variables into Principal Components,” Journal of Educational Psychology, vol. 24, pp. 417-441, 498-520, 1933. 27. T. Hastie and W. Stuetzle, “Principal Curves,” Journal of the American Statistical Association, vol. 84, pp. 502-516, 1989. 28. D. Asimov, “The Grand Tour: A tool for Viewing Multidimensional Data,” DIAM Journal on Scientific and Statistical Computing, vol. 61, pp. 128-143, 1985. 29. J. H. Friedman, “Exploratory Projection Pursuit,” Journal of the American Statistical Association, vol. 82, pp. 249-266, 1987. 30. T. Kohonen, E. Oja, O. Simula, A. Visa, and J. Kangas, “Engineering Applications of the Self-Organizing Map,” presented at IEEE, 1996. 31. G. Grinstein, P. E. Hoffman, S. Laskowski, and R. Pickett, “Benchmark Development for the Evaluation of Visualization for Data Mining,” in Information Visualization in Data Mining and Knowledge Discovery, The Morgan Kaufmann Series in Data Managament Systems, U. Fayyad, G. Grinstein, and A. Wierse, Eds., 1st ed: Morgan-Kaufmann Publishers, 2001. 32. Voigt, K. and Bruggeman, R. (1995) Toxicology Databases in the Metadatabank of Online Databases Toxicology, 100, 225-240 33. Weinstein, J.N.,et.al., (1997,) An information-intensive approach to the molecular pharmacology of cancer, Science, 275, 343-349. 34. Shi, L.M., Fan, Y.,Lee, J.K., Waltham, M., Andrews, D.T., Scherf,U., Paul, K.D., and Weinstein, J.N. (2000) J. Chem. Inf. Comput. Sci., 40, 367-379. 35. Bai, R.L., Paul, K.D., Herald, C.L., Malspeis, L., Pettit, G.R., and Hamel, E. (1991) Halichondrin B and homahalichondrin B, marine natural products binding in the vinca domain of tubulin-based mechanism of action by analysis of fifferential cytotoxicity data J. Biol. Chem., 266, 15882 – 15889. 36. Cleveland, E.S., Monks, A., Vaigro-Wolff, A., Zaharevitz, D.W., Paul, K., Ardalan, K.,Cooney, D.A., and Ford, H. Jr. (1995) Site of action of two novel pyramidine biosynthesis inhibitors accurately predicted by COMPARE program Biochem. Pharmacol., 49, 947-954. 37. Gupta, M., Abdel-Megeed M., Hoki, Y, Kohlhagen, G., Paul, K., and Pommier, Y. (1995) Eukaryotic DNA topoisomerases mediated DNA cleavage induced by new inhibitor: NSC 665517 Mol. Pharmacol., 48, 658-665 38. Shi, L.M., Myers, T.G., Fan, Y., O’Connors, P.M., Paul, K.D., Friend, S.H., and Weinstein, J.N. (1998) Mining the National Cancer Institute Anticancer Drug Discovery Database: cluster analysis of ellipticine analogs with p53-inverse and central nervous system-selective patterns of avtivity Mol. Pharmacology, 53, 241-251. 39. Ross, D.T. et. al., (2000) Systemamtic variation of gene expression patterns in human cancer cell lines Nat. Genet., 24, 227-235 40. Staunton, J.E.; Slonim, D.K.; Coller, H.A.; Tamayo, P.; Angelo, M.P.; Park, J.; Sherf, U.; Lee, J.K.; Reinhold, W.O.; Weinstein, J.N.; Mesirov, J.P.; Landers, E.S.; Golub, T.R. Chemosensitivity prediction by transcriptional profiling, Proc. Natl. Acad. Sci., 2001, 98, 10787-10792. 41. Marx, K.A., O’Neil, P., Hoffman, P.; Ujwal, M.L. Data Mining the NCI Cancer Cell Line Compound GI50 Values: Identifying Quinone Subtypes Effective Against Melanoma and Leukemia Cell Classes, J. Chem. Inf. Comput. Sci., 2003, in press. 42. Blower, P.E.; Yang, C.; Fligner, M.A.; Verducci, J.S.; Yu, L.; Richman, S.; Weinstein, J.N. Pharmacogenomic analysis: correlating molecular substructure classes with microarray gene expression data, The Pharmacogenomics Journal, 2002, 2, 259271. 43. Scherf, W.; Ross, D.T.; Waltham, M.; Smith, L.H.; Lee, J.K.; Tanabe, L.; Kohn, K.W.; Reinhold, W.C.; Myers, T.G.; Andrews, D.T.; Scudiero, D.A.; Eisen, M.B.; Sausville, E.A.; Pommier, Y.; Botstein, D.; Brown, P.O.; Weinstein, J.N. A gene expression database for the molecular pharmacology of cancer, Nature, 2000, 24, 236247. 44. Schafer, J.L. Analysis of Incomplete Multivariate Data, Monographs on Statistics and Applied Probability 72, Chapman & Hall/CRC, 1997. 45. RadViz, URL: www.anvilinfo.com 46. Hoffman, P.; Grinstein, G.; Marx, K.; Grosse, I.; Stanley, E. DNA visual and analytical data mining, IEEE Visualization 1997 Proceedings, pp. 437-441, Phoenix 47. Hoffman, P.; Grinstein, G. Multidimensional information visualization for data mining with application for machine learning classifiers, Information Visualization in Data Mining and Knowledge Discovery, Morgan-Kaufmann, San Francisco, 2000. 48. Bucci, C.; Thompsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: a key to lysosome biogenesis, Mol. Biol. Cell, 2000, 11, 467-480. 49. Ross, D. NAD(P)H: quinone oxidoreductases, Encyclopedia of Molecular Medicine, 2001, 2208-2212. 50. Ross, D.; Beall, H.; Traver, R.D.; Siegel, D.; Phillips, R.M.; Gibson, N.W. Bioactivation of quinones by DT-Diaphorase. Molecular, biochemical and chemical studies, Oncology Research, 1994, 6, 493-500 51. Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structure of recombinant human and mouse NAD(P)H:quinone oxidoreductase: Species comparison and structural changes with substrate binding and release, Proc. Natl. Acad. Sci., 2000, 97, 3177-3182 52. Faig, M.; Bianchet, M.A.; Winsky, S.; Moody, C.J.; Hudnott, A.H.; Ross, D.; Amzel, L.M. Structure-based development of anticancer drugs: complexes of NAD(P)H:quinone oxidoreductase 1 with chemotherapeutic quinones, Structure (Cambridge), 2001, 9, 659667 53. Wolkenberg, S.E. In situ activation of antitumor agents, Tetrahedron Lett., 2001, 1-5 54. Smith, M.T.; Wang, Y.; Kane, E.; Rollinson, S.; Wiemels, J.L.; Roman, E.; Roddam, P.; Cartwright, R.; Morgan, G., Low NAD(P)H: quinone oxidoreductase I activity is associated with increased risk of acute leukemia in adults, Blood, 2001, 97, 1422-1426 55. Wiemels, J.L.; Pagnamenta, A.; Taylor, G.M.; Eden, O.B.; Alexander, F.E.; Greaves, M.F. A lack of a functional NAD(P)H:quinone oxidoreductase allele in selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators, Cancer Res., 1999, 59, 4095-4099 56. Naoe T.; Takeyama, K.;, Yokozawa, T.; Kiyoi, H.; Seto, M.; Uike, N.; Ino, T.; Utsunomiya, A.; Maruta, A.; Jin-nai, I.; Kamada, N.; Kubota, Y.; Nakamura, H.; Shimazaki, C.; Horiike, S.; Kodera, Y.; Saito, H.; Ueda, R.; Wiemels, J.; Ohno, R. Analysis of the genetic polymorphism in NQO1, GST-M1, GST-T1 and CYP3A4 in 469 Japanese patients with therapy related leukemia/myelodysplastic syndrome and de novo acute myeloid leukemia, Clin. Cancer Res., 2000, 6, 4091-4095 Other References (14-25 in CC Grant) 35. Venter, J.C., et.al., The Sequence of the Human Genome. Science, 291, 1303-1351 (2001). 36. Lander, E.S., et.al., Initial Sequencing and Analysis of the Human Genome. Nature, 409, 860-921 (2001). 37. Stoeckert, C.J., et.al., Microarray databases: standards and ontologies. Nat. Genet. 32 (Suppl) 469-473. 38. No author, Microarray standards at last. Nature, 419, 323. 39. Ball, C., et.al., Standards for microarray data., Science, 298, 539. 40. Quackenbush, J. (2001) Computational analysis of cDNA microarray data. Nature Reviews 2(6): 418-428. 41. Dudoit, S., Yang, Y.H., Speed, T.P., and Callow, M.J. (2002) Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica Vol. 12, No. 1, p. 111-139. 42. Li, C. and Wong, W.H. (2001) Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error applications. Genome Biology 2(8), 43. Irizarry, R.A., Hobbs, B., Collin, F., Beazer-Barclay, Y.D., Antonellis, K., Scherf, U., and Speed, T.P. (2003) Exploration, normalization and summaries of high density oligonucleotide array probe level data. Biostatistics (in press). 44. Durbin, B.P., Hardin, J.S., Hawkins, D.M., and Rocke, D.M. (2002) A variancestabilizing transformation for gene expression microarray data. Bioinformatics 18, 105S110S. 45. Bolstad, B.M., Irizarry, R.A., Astrand, M., and Speed, T.P. (2002) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19(2): 185-193. Schadt, E.C., Li, C., Eliss, B., and Wong, W.H. (2002) Feature extraction and normalization algorithms for high-density oligonucleotide gene expression array data. J. Cell. Biochem. 84(S37), 120-125. Figure Legends Figure 1. RadViz figure Figure 2. Cancer cell line functional class definition using a hierarchical clustering (1Pearson coefficient) dendrogram for 60 cancer cell lines based upon gene expression data. Five well defined clusters are shown highlighted. We treat the highlighted cell line clusters as the truth for the purpose of carrying out studies to identify which chemical compounds are highly significant in their classifying ability Figure 3. RadViz™ result for the 3-Class problem classification of melanoma, leukemia and non-melanoma, non-leukemia cancer cell types at the p < .01 criterion. Cell lines are symbol coded as described in the figure. A total of 14 compounds (bottom of layout) were most effective against melanoma and they are layed out on the melanoma sector (counterclockwise from most to least effective). For leukemia, 30 compounds were identified as most effective and are layed out in that sector. Some 8 compounds were found to be most effective against non-melanoma, non-leukemia cell lines and are layed out in that sector. Figure 4. One example each of the two quinone subtypes selected in Figure 3 are displayed. A. The most highly effective of the 11 internal quinone subtype compounds most effective against melanoma is shown. B. The most highly effective of the 6 external quinone subtype compounds most effective against leukemia is shown Figure 5. RadViz™ result for the 3-Class Problem classifying the following three classes: acute lymphoblastic leukemia (ALL), non-ALL leukemia (other-Leukemia) and non-leukemia cell classes at p < .05. We used as input the 30 compounds identified in the Figure 3 classification as most effective against all leukemias at the p < .01 selection criterion. Cell lines are symbol coded as described in the figure. The NSC numbers of the compounds selected to classify the classes are presented in the order of their ranking from most effective to least effective moving counterclockwise within each class sector. 0.2 ME_LOXIMVI PR_PC-3 PR_DU-145 RE_SN12C 0.6 0.0 LC_HOP-92 BR_MDA-MB-231/ATCC CNS_SF-295 CNS_SNB-19 CNS_U251 BR_BT-549 CNS_SF-268 CNS_SF-539 CNS_SNB-75 BR_HS578T RE_A498 RE_CAKI-1 RE_ACHN RE_UO-31 RE_TK-10 RE_RXF-393 RE_786-0 LC_NCI-H226 LC_HOP-62 OV_OVCAR-8 BR_MCF7/ADF-RES LC_NCI-H23 LC_NCI-H522 LC_NCI-H460 LC_A549/ATCC LC_EKVX LE_SR LE_RPMI-8226 LE_K-562 LE_HL-60 LE_CCRF-CEM LE_MOLT-4 OV_SK-OV-3 OV_IGROV1 OV_OVCAR-3 OV_OVCAR-4 OV_OVCAR-5 LC_NCI-H322M BR_MCF7 BR_T-47D CO_HCT-116 CO_SW-620 CO_HCT-15 CO_KM12 CO_HT29 CO_HCC-2998 CO_COLO205 BR_MDA-MB-435 BR_MDA-N 0.4 ME_SK-MEL-5 ME_MALME-3M ME_SK-MEL-28 ME_UACC-257 ME_SK-MEL-2 ME_UACC-62 ME_M14 Height Cluster Dendrogram 1.0 0.8 O O O N+ - O O O O O O N O N H N H H Cl N H N O O O S N O N H N NH O O 1. 670762 N+ Cl 2. 670766 O 3. 642061 O O O O H N O H O S S N N _ O O H 4. 658450 5. 602617 6. 690432 O O S Cl O S O H O N O O O Cl 8. 644902 7. 690434 O S O H S S O 10. 656239 O O N O S N O 11. 628507 O 9. 642009 H H O A O N O O O N O O O O H 3. 618315 2. 641395 1. 648147 O O O O S N O O O 4. 641394 5. 640192 B O Cl O N H O 1. 621179 N O 6. 641396