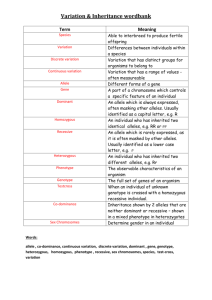
Variation:
advertisement

MOLECULAR GENETICS 2005 Tuesday November 29: Human Genetics, part I Liisa Kauppi (Keeney lab) RRL-1129 Phone 639 5180 Email kauppiL@mskcc.org Web resources: For BLAST searches, gene queries etc.: http://www.ensembl.org/Homo_sapiens/index.html For comprehensive listing and information on genetic diseases: Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM SNP database: http://www.ncbi.nlm.nih.gov/SNP/index.html HapMap project: http://www.hapmap.org/index.html.en Textbook: Human molecular genetics (Strachan and Read), chapters 11 and 12 in version 2 of the book See any recent issue of American Journal of Human Genetics for applications of the methods described in this lecture (at www.ajhg.org). Human genetics can be studied in pedigrees or populations. There are limitations, however: unlike with model organisms such as mice or flies, in human genetics you cannot generate mutants, you cannot perform controlled matings, place your subjects in a controlled environment etc. We have to rely on “natural” mutants, i.e. patients with a phenotype. For a trait to be amenable to genetic analysis, it has to show some degree of heritability. This can be assessed in pedigrees (first degree relatives, twins). There are two basic types of genetic diseases: simple Mendelian with complete correlation between genotype and phenotype - if you have the mutant gene, you'll get the disease, and complex or multifactorial (risk of getting the disease is modified by individual's genotype; other factors like other genes and environment, also influence disease risk). Mapping disease genes in humans is done by using polymorphic DNA markers (single nucleotide polymorphisms [SNPs] and microsatellites). Terminology for SNPs is defined by allele frequency. A single base change, occurring in a population at a frequency of >1% is termed a SNP. When a single base change occurs at <1% it is considered to be a mutation. Expected genotype frequencies at any bi-allelic locus can be calculated using the HardyWeinberg equation. It has the following assumptions: large randomly mating population, no mutations, no migration between populations, no selection (all genotypes reproduce with equal success). Basic relations are: two alleles at a locus (B and b) frequency of the B allele = p, frequency of the b allele = q and p + q = 1 Genotype frequencies are given by the equation: p2 (BB) + 2pq (Bb) + q2 (bb) = 1 Any individual is the result of the union of two gametes which have the probability of containing a specific allele (equal to the allele frequency). This can be diagrammed as a Punnett square: p (B) q (b) p (B) p2 (BB) pq (Bb) q (b) pq (Bb) q2 (bb) HWE explains how recessive traits are maintained in a population, and allows calculations of carrier frequencies for recessive traits (with some caution). Departure from HWE suggests that some assumptions are not met, for example there may be recent migration or selection influencing the genotype frequencies. Family studies In a simple Mendelian disease, polymorphisms are studied in family members for genetic linkage. If the marker is close to the disease gene on the chromosome there is a low chance of recombination at meiosis and linkage is observed; if the marker and disease gene are far apart (or on different chromosomes), linkage is not observed. To calculate linkage, the number of recombinant (R) and non-recombinant (NR) offspring are counted for each sibship in the family. If the recombination fraction RF (), calculated as R/(R+NR), is significantly less than 0.5, there is evidence of linkage. Numbers of offspring in human families are small. In order to get statistically significant evidence for linkage, it is necessary to combine evidence from many families, and to make probabilistic guesses for families where not all members are available for study (all done on a computer). The lod (logarithm of odds) score Z is a statistic that describes the strength of evidence for linkage, at any chosen value of the RF, given the family data available. It is calculated as follows: Z= log [Likelihood of loci being linked/Likelihood of loci not being linked] A lod score of 3 or more is considered good evidence for linkage and a lod score of -2 or less is evidence against linkage. Values between -2 and 3 are inconclusive and more data must be obtained. A genome scan consists of genotyping a collection of families with the genetic disease using hundreds markers across the genome. By using many markers there is a good chance that any unknown gene will be close enough to one or two of them to show genetic linkage. The aim is to find linkage with two markers, one of which is on each side of the disease gene; then the disease gene must be between the two markers (usually a few Mb apart). Recognizing recombinants does the disease segregate with this marker? 1 I 25 16 II 6 21 34 III 31 32 41 NR NR NR 41 NR 42 32 NR R Recombination fraction is 1/6=0.167 Which marker is the disease locus closest to? Multi-point lod scores chr 3p12-14 Waardenburg syndrome type 2 Hughes et al. (1994) Nature Genet 7, 509-512 “Shared segment” analyses When there is no simple Mendelian inheritance pattern, we can analyze affected people only. When you only look at affecteds, there is no need to specify penetrance of the disease. In affected sib pair analysis, the aim is to identify genomic segments that are shared between the affected sibs more often than expected by chance. Association studies in populations In populations, cases (patients) and controls can be collected. Then you look for alleles that are more prevalent in the patient cohort than in the general population (controls). However, typing many markers in such large numbers of samples would be prohibitively expensive. Luckily, we can make use of a phenomenon called linkage disequilibrium (LD). This refers to the correlation between two alleles on a chromosomal segment. LD is a result of this chromosomal segment (haplotype) not having been broken up by meiotic recombination in the population. A haplotype is the set of alleles at more than one locus that is inherited by an individual from one of its parents. For two loci with two alleles each (A and a, B and b) there are four possible haplotypes: AB, Ab, aB, ab. A diploid individual’s genotype could be for example AB on one homologous chromosome and Ab on the other homolog. In other words, the individual has two haplotypes, one inherited from each parent, just as a one-locus genotype contains two alleles from the two parents. The opposite of LD is linkage equilibrium (or free association), which can be thought of as the two-locus version of the Hardy-Weinberg ratio, but it is a property of haplotypes, not genotypes. At linkage equilibrium, all four haplotypes are found in the population, at frequencies that are expected based on the genotype frequencies of the individual alleles. Linkage disequilibrium (LD) measures association between two alleles Mutation creates new variants A G A A A G T A Initially, the new allele is in LD with nearby alleles LD value = 1 Recombination reshuffles existing variation A G A G X T A A T LD diminishes If enough crossovers take place, the loci are in Тfree associationУ Commonly used LD measures: D Хand r2 The utility of LD for gene mapping is the fact that if there are extended chromosomal segments where alleles are inherited as “a package” (haplotype blocks), then there is no need to type every SNP in such a segment. It should be sufficient to select a few “tag SNPs” for a haplotype block and only genotype those. Recently, the HapMap project has characterized the extent of haplotype blocks in four human populations (Nigerians, Whites in the US, Japan, China), and identified SNPs for tagging these blocks. LD-based methods work best when there is a single susceptibility allele at any given disease locus, and generally perform very poorly if there is substantial allelic heterogeneity. The Common-Disease Common-Variant (CDCV) hypothesis states that disease-predisposing variants exist at relatively high frequency (i.e. >1%) in the population. According to the CDCV hypothesis, common diseases are caused by ancient alleles occurring on specific haplotypes; these haplotypes should be detectable in a casecontrol study using tagging SNPs. The alternative (and more pessimistic) hypothesis states that disease-predisposing alleles are sporadic new mutations, perhaps around the same genes, on different haplotypes. Different families with history of the same disease would then owe their condition to different mutations events. There are examples to show that for at least some common diseases the CDCV hypothesis is true (Alzheimer’s etc.). It is likely, however, that some diseases have underlying allelic heterogeneity. For any gene, there are a number of ways in which SNPs can alter its expression (nonsynonymous, modify splicing, alter promoter function etc.). If a pathway leading to the disease is complex (consider schizophrenia for example), the number of genes involved is higher, and the likelihood of several SNPs affecting gene function increases. Finally, recently it has been discovered that all humans carry several low copy number polymorphisms in their genomes (see Mike Wigler’s President’s seminar talk on Dec 14). It is likely that these too will influence susceptibility to common disease. There is also evidence that microsatellite and insertion/deletion (indel) polymorphisms can influence phenotypes: - Spinocerebellar ataxia Type10 (SCA10) (OMIM:+603516) is caused by the largest tandem repeat seen in human genome. Normal population has 10-22 repeats of the pentanucleotide ATTCT in intron 9 of SCA10 gene; SCA10 patients have 800-4500 repeat units that cause the disease allele to be up to 22.5 kb larger than the normal one. - Association between coronary heart disease and a 287-bp indel polymorphism located in intron 16 of the angiotensin converting enzyme (ACE) has been reported (OMIM 106180). This indel, known as ACE/ID is responsible for 50% of inter-individual variability of plasma ACE concentration.