pro2792-sup-0001-suppinfo
advertisement

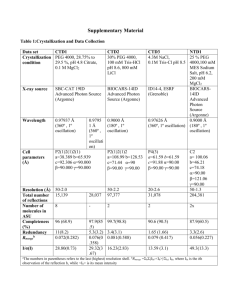
Supplementary text: Relationship between Conservation of Interface Location (CIL) and Conservation of Interaction pattern (CIP) scores and binding affinity for transient complexes Although transient protein-protein complexes dissociate and associate to carry out biological functions, their binding affinities are variable; there can be strong transient interactions or weak transient interactions. The strong interactions typically require a trigger for association / dissociation, whereas the weak interactions show a fast bound – unbound equilibrium1. For the main dataset of 93 transient complex pairs used in this study (Table 1, Table 2), binding affinity values were culled from two main curated sources documenting binding affinity for complexes of known structure based on manual curation: structure-based benchmark for protein binding affinity2, PDBbind database v20143. Experimentally measured binding affinity information is available for at least 1 complex in the pair in 56% cases (n=53), with values available for both complexes in 12 cases (for 50%, the binding affinity values of homologous complexes are very similar). The pairs were divided into 3 sets: Low affinity (Kd ≤ 10-7 M, n = 10), Low-Medium affinity (Kd ≤ 10-8 M, n = 31), and High affinity pairs (Kd ≥ 10-9 M, n = 22). The variation between the binding affinity of transient complexes and CIL and CIP scores was analysed (Supplementary Figure S4). The scatter plots and box plots indicate that there is no correlation between the measures. However, this result should be viewed in light of some mitigating factors: a) Experimentally reported binding affinity values can vary commonly by a factor of 2 even when studied under similar conditions of pH, temperature, ionic strength2. b) There are only few data points available for low affinity complexes (in the micromolar range) In summary, there appears to be no distinct trend for CIL and CIP scores in strong and weak transient binders. However, larger amounts of data perhaps in the future is essential to confirm this result. An approach to improve comparative modelling of protein-protein complexes using multiple templates: A case study with GTPase-GAP complex. Mouse GTPase-GAP complex was modelled starting from the tertiary structures and the accuracy of the modelled structure has been assessed in comparison to the crystal structure of the complex (pdb id: 3c7k)4. In order to build a model, information about residue pairs with conserved interaction pattern was obtained from six homologous templates of mouse GTPaseGAP complex (pdb id: 1agr, 1fqj, 2gtp, 2ik8, 2ihb, 2ode) (Supplementary figure S6a).These residues were used for guided protein docking using HADDOCK program5 (refer Materials and Methods). The assessment of the modelled complex is made by comparing the overlap of interface residues and the interacting pairs as identified in the crystal structure (pdb: 3c7k). The docked model was able to correctly predict 88% of the interactions in the protein-protein complex (15 out of 17 interactions from the crystal structure. 11 out of 12 interface residues were predicted for one chain and all the 12 interface residues were predicted for the other chain. 7 residue pairs have been wrongly predicted as interacting residues with some of them being interface residues). These encouraging results led us to use information of residues with conserved interaction pattern only from one template of GTPase-GAP complex (pdb id: 2ihb) (Supplementary figure S6b) whose conservation of interaction pattern score is only 48.5%. Docking results showed equally good results of picking up 76% of the correct interactions in the protein-protein complex (This includes 13 correct interactions out of 17 total numbers of interactions. 10 out of 13 interface residues in one chain and 11 out of 12 interface residues in the other chain were correctly predicted. 6 residue pairs were wrongly predicted to be interacting pairs). It was found that the consensus interaction from all the 12 best docked models showed high confidence for the correctly predicted interactions and low confidence for majority of the wrongly predicted interactions. Moreover, three of them have poor conservation score emphasizing the wrong prediction. We wanted to assess the accuracy of the models thus obtained by supposedly improved method with that of the model obtained using more simple-minded traditional method. Therefore GTPase-GAP complex was also modelled by the traditional method of superimposing the tertiary structure of the target chains onto the template complex using DALI to directly extrapolate the interface residues and interacting residue pairs to the complex to be modelled from the template. The number of correctly predicted interface residues and interacting pairs are lower than CIP residue guided protein docking. Only 59% of interactions were correctly predicted by this method (10 interacting pairs were correctly predicted out of 17. This includes 7 interface residues out of 13 in one chain and 7 out of 12 residues from the other chain. The number of wrongly predicted interface residues is 7). It should be noted that all the core-core interactions in these homologous pairs of protein complexes showed conserved interaction pattern. This test case shows that there is lot of scope to integrate this method with other features using machine learning algorithms6 to improve the accuracy of homology modelled protein-protein complexes. References 1. Kastritis PL, Moal IH, Hwang H, Weng Z, Bates PA, Bonvin AM, Janin J. A structurebased benchmark for protein-protein binding affinity. Protein Sci 2011; 20: 482-91. 2. Wang R, Fang X, Lu Y, Wang S.The PDBbind database: Collection of binding affinities for protein-ligand coplexes with known three-dimensional structures. J Med Chem 2004; 47: 2977-80. 3. Perkins, J.R., Diboun, I., Dessailly, B.H., Lees, J.G. and Orengo, C. Transient proteinprotein interactions: structural, functional, and network properties. Structure, 18, 12331243. 4. Slep KC, Kercher MA, Wieland T, Chen CK, Simon MI, et al. Molecular architecture of Galphao and the structural basis for RGS16-mediated deactivation. Proc Natl Acad Sci U S A 2008; 105: 6243-6248. 5. Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 2003; 125: 1731-1737. 6. Koike A, Takagi T. Prediction of protein-protein interaction sites using support vector machines. Protein Eng Des Sel 2004; 17: 165-173. Supplemental information Legends to Supplementary figures Figure S1 (a) Correlation between the extent of conservation of interface location (CIL) for both homologous subunits (AA’ and BB’) of pairs of transient Protein-Protein Complex (PPC). Lowest and highest CIL scores are plotted in X-axis and Y-axis respectively. (b) Histogram of CIL scores for a dataset of same protein complexed to homologous proteins. (c) Eglin C bound to homologous proteins subtilisin carlsberg (PDB code: 1cse) and thermitase (PDB code: 1tec) are coloured as dark and light green. Residues with CIL are shown as pink sticks. Both the pairs of homologous subunits of the protein complex shows high CIL score. (d) Box plot representation for the distribution of conservation of interface location for identical pairs of transient PPC and permanent homodimers solved in different crystal conditions. (e) Correlation between CIL and global sequence identity for homologous pairs of transient PPC and permanent homodimers. Figure S2 Correlation between conservation of interface location (CIL) and extent of structurally equivalent conserved residue type at interfaces of homologous pairs of transient PPC and permanent dimers. Figure S3 (a) Distribution of extent of conservation of interaction pattern (CIP) for a dataset of same protein complexed to homologous proteins. (b) Eglin C bound to homologous proteins subtilisin carlsberg (1cse) and thermitase (1tec) are coloured in dark and light green. Residues with CIP are shown as pink sticks and the common interactions are shown as dotted lines. The homologous pair of the PPCs shows high CIP score. (c) Correlation between CIL and CIP for homologous pairs of transient PPC and permanent homodimers. Figure S4 The variation of CIL (Conservation of Interface Location) and CIP (Conservation of Interaction Pattern) scores across affinity between interacting proteins in the complexes. Scatter plots of (a) Average CIL score (calculated as CIL1 + CIL2 / 2) and log(Binding Affinity) and (b) CIP score and log(Binding Affinity) are shown, indicating no correlation. Further, subdividing the complexes into groups of low affinity (≤10-7M Kd, n=10), low+medium affinity (≤10-8 M Kd, n=31), and high affinity (≥10-9 M Kd, n=22) complexes and comparing their (c) Average CIL scores (d) CIP scores using Wilcoxon test shows that there is no statistically significant correlation. Similar analyses carried out for Min (CIL1,CIL2) and Max (CIL1,CIL2) gave similar results. Figure S5 (a) Extent of conservation of interface location (CIL) for core and rim interface residues for the dataset of same protein bound to homologous proteins. (b) Extent of CIL for core and rim interface residues with conserved residue type are shown for each of the twenty residue types (c) Extent of conservation of interaction pattern (CIP) for core-core, core-rim and rim-rim interaction categories are shown. Figure S6 (a) Residues showing conserved interaction pattern (CIP) from multiple templates are provided to guide the docking of GTPase-GAP complex (PDB id: 3c7k). The interacting chains are coloured in dark and light grey. Correctly and wrongly predicted interface residues are coloured in green and red respectively. (b) Similarly, residues showing conserved interaction pattern (CIP) from a single template (PDB id: 2ihb) is provided to guide the docking of the GTPase-GAP complex (PDB id: 3c7k). Supplemental information Legends to Supplementary figures Figure S1 (a) Correlation between the extent of conservation of interface location (CIL) for both homologous subunits (AA’ and BB’) of pairs of transient Protein-Protein Complex (PPC). Lowest and highest CIL scores are plotted in X-axis and Y-axis respectively. (b) Histogram of CIL scores for a dataset of same protein complexed to homologous proteins. (c) Eglin C bound to homologous proteins subtilisin carlsberg (PDB code: 1cse) and thermitase (PDB code: 1tec) are coloured as dark and light green. Residues with CIL are shown as pink sticks. Both the pairs of homologous subunits of the protein complex shows high CIL score. (d) Box plot representation for the distribution of conservation of interface location for identical pairs of transient PPC and permanent homodimers solved in different crystal conditions. (e) Correlation between CIL and global sequence identity for homologous pairs of transient PPC and permanent homodimers. Figure S2 Correlation between conservation of interface location (CIL) and extent of structurally equivalent conserved residue type at interfaces of homologous pairs of transient PPC and permanent dimers. Figure S3 (c) Distribution of extent of conservation of interaction pattern (CIP) for a dataset of same protein complexed to homologous proteins. (b) Eglin C bound to homologous proteins subtilisin carlsberg (1cse) and thermitase (1tec) are coloured in dark and light green. Residues with CIP are shown as pink sticks and the common interactions are shown as dotted lines. The homologous pair of the PPCs shows high CIP score. (c) Correlation between CIL and CIP for homologous pairs of transient PPC and permanent homodimers. Figure S4 The variation of CIL (Conservation of Interface Location) and CIP (Conservation of Interaction Pattern) scores across affinity between interacting proteins in the complexes. Scatter plots of (a) Average CIL score (calculated as CIL1 + CIL2 / 2) and log(Binding Affinity) and (b) CIP score and log(Binding Affinity) are shown, indicating no correlation. Further, subdividing the complexes into groups of low affinity (≤10-7M Kd, n=10), low+medium affinity (≤10-8 M Kd, n=31), and high affinity (≥10-9 M Kd, n=22) complexes and comparing their (c) Average CIL scores (d) CIP scores using Wilcoxon test shows that there is no statistically significant correlation. Similar analyses carried out for Min (CIL1,CIL2) and Max (CIL1,CIL2) gave similar results. Figure S5 (a) Extent of conservation of interface location (CIL) for core and rim interface residues for the dataset of same protein bound to homologous proteins. (b) Extent of CIL for core and rim interface residues with conserved residue type are shown for each of the twenty residue types (c) Extent of conservation of interaction pattern (CIP) for core-core, core-rim and rim-rim interaction categories are shown. Figure S6 (a) Residues showing conserved interaction pattern (CIP) from multiple templates are provided to guide the docking of GTPase-GAP complex (PDB id: 3c7k). The interacting chains are coloured in dark and light grey. Correctly and wrongly predicted interface residues are coloured in green and red respectively. (d) Similarly, residues showing conserved interaction pattern (CIP) from a single template (PDB id: 2ihb) is provided to guide the docking of the GTPase-GAP complex (PDB id: 3c7k).