RED BLOOD CELLS DISORDERS: ANEMIAS, POLYCYTEMIA

RED BLOOD CELLS DISORDERS: ANEMIA, POLYCYTEMIA. BLEEDING
DISORDERS. prof. Alena Skálová, MD, Ph
REVIEW OF STRUCTURE AND FUNCTIONS OF BLOOD ELEMENTS peripheral blood plasma-fluid in which the formed elements of the blood are suspended
-plasma is composed of water, electrolytes, proteins (albumin, globulins), glucose, enzymes, etc.
-measurement of different plasma constituents may provide evidence of disease serum- is the fluid that remains after blood has been allowed to clot
-it resembles plasma, except of the fact that fibrinogen and other coagulation factors have been used (depleted) by the process of clot formation
RBCs (erythrocytes)- nonnucleated biconcave cells with uniform diameter of 8
µm
-contain hemoglobin, a protein complexed to the iron containing porphyrin
-hemoglobin is a vital to oxygen transport
-life span of RBCs is 120 days
-RBCs are derived from normoblasts, they occur in the bone marrow, never in peripheral blood reticulocytes- are newly released immature RBCs (1 % in peripheral blood) white blood cells (granulocytes)- nucleated white blood cells, classified according to the nature of their cytoplasmic granules as - neutrophils, eosinophils, basophils myelopoiesis in the bone marrow includes: stem cells-myeloblast-promyelocyte-myelocyte-fully mature granulocyte (4-5 nuclear lobes) normal bone marrow structure
-bone marrow is the site of hematopoiesis at birth- active hematopoietic bone marrow is present in medullary cavities of all bones at adult age- hematopoietic bone marrow is partly replaced by adipose tissue in the bones of extremities, active BM is present only in the axial skeleton (ribs, vertebrae, iliac crest, sternum) examination of the bone marrow in adults- specimens must be obtained from the axial bones, most commonly from sternum- as aspiration biopsy- utilizes a thin needle and provides a material for smears and for histologic sections iliac crest- as trephine biopsy- utilizes larger needle that provides a core of tissue ( including the bone spicules) for histologic sections
1
in the bone marrow specimen- we evaluate various features, such as:
-amount and maturation of erythropoiesis and myelopoiesis
-number and morphology of megakaryocytes
-number and morphology of plasma cells
-presence of tumor cells- infiltration by metastatic carcinoma, malignant lymphoma, leukemias, multiple myeloma
-presence of fibrosis, iron deposits, regressive changes, lymphocytic infiltration, amount of adipose component, etc.
ANEMIA
= is reduction in the oxygen transport capacity of blood, usually due to reduction of total amount of circulating red cells
anemia is characterized by lower than normal hematocrit = ratio of erythrocytes to plasma- normal 40:60, normal hematocrit is about 42-
50%,
hematocrit is an important determinant of blood viscosity
-lower than normal hemoglobin concentrations
-increased erythropoiesis- due to increased production of erythropoetinin most anemias-causes erythroid bone marrow hyperplasia, increased erythropoiesis may also occur in the spleen and liver -sites of possible extramedullary hemopoiesis
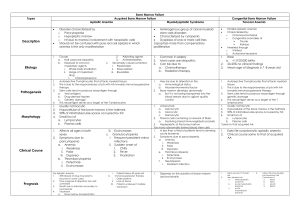
CLASSIFICATION OF ANEMIA -is based on the mechanism
I. anemia due to decreased erythropoiesis
II. anemia of blood loss (acute and chronic)
III. anemia due to increased rate of destruction of erythrocytes (hemolytic anemia)
1./ intrinsic (intracorpuscular ) abnormalities of RBCs –hereditary and acquired
2./ extrinsic (extracorpuscular) abnormalities
Common clinical features of all anemias:
-characterized by pallor of the skin and mucous membranes
-by symptoms that represent a manifestation of hypoxia (=decreased oxygen content of the blood )- fatigue, breathlessness
-myocardial hypoxia may result in angina pectoris with chest pain, or even cardiac failure
-decreased hemoglobin level identification of the type of anemia - requires examination of peripheral blood and sometimes bone marrow specimen
2
ad I./ ANEMIAS DUE TO DECREASED ERYTHROPOIESIS
-impaired RBCs production may be caused by a variety of disorders major pathogenetic groups of anemia caused by a decreased erythropoiesis
1. Aplastic anemias
2. Anemias caused by bone marrow replacement
3. Anemias due to defective synthesis of DNA
4. Anemias due to defective hemoglobin synthesis
1./ APLASTIC ANEMIA
-is due to failure of production, or due to suppression or destruction of stem cells in BM, which leads to decreased generation of RBCs- anemia failure of production of RBCs, granulocytes and platelets= pancytopenia
BM shows decrease of cellularity etiology:
-may be idiopathic - no identifiable cause
-drugs- is by far the most common known cause of aplastic anemia- damage to the BM may be dose-related and predictable (alkylating agents, antimetabolites, such as vincristin, busulfan) or idiosyncratic and thus unpredictable, such as caused by chloramphenicol, chlorpromazine, streptomycin
-irraditaion
-some infections or inherited diseases, like Fanconi anemia, which is associated with multiple congenital anomalies pathogenesis:
-in idiopathic cases, the stem cell failure may be due to primary structural defect of stem cells
-or mediated by immune-suppression of the stem cells, then may be reversed by immunosuppression therapy morphology:-bone marrow is hypocellular - active BM is replaced by fat cells clinically:
-all symptoms are related to pancytopenia
-leading feature is usually bleeding diathesis
PURE RED CELL APLASIA- very rare form of BM failure
-results from isolated absence of RBC precursors
-in acute form- may be related to drug administration or to viral infection
-may occur as a complication of thymoma (by an unknown mechanism)
3
2./ ANEMIAS DUE TO BM REPLACEMENT a) involvement of the bone marrow cavity by malignant tumors
-most often - in leukemias - malignant neoplastic proliferations of hematopoietic cells
-infiltration of BM by lymphoma - neoplastic proliferation derived of lymphoid cells arising most often in lymph nodes
-infiltration by metastatic carcinomas, such as breast ca, bronchogenic ca, etc-less common
Types of BM involvement:
-focal - ca, lymphoma, multiple myeloma
-interstitial - some types of leukemia, non-hematological malignancies
-diffuse- most leukemias b) replacement of the BM by fibrosis
-most importantly in osteomyelofibrosis compensatory phenomenon in all types of BM replacements include
-extramedullary hematopoiesis - development of active marrow in sites outside the bone marrow cavity- (spleen, liver)
-release of cells from the marrow before maturation is complete
-results in „shift to left„- implies a release of immature white cells including the band forms and even of myeloblasts and myelocytes and normoblasts (nucleated
RBCs ) into the peripheral blood
3./ ANEMIAS DUE TO DEFECTIVE SYNTHESIS OF DNA=
MEGALOBLASTIC ANEMIAS
megaloblastic anemias - are characterized by disorder in maturation phase of erythropoiesis resulting in formation of abnormal erythroid precursors which are enlarged and show failure of maturation - these are called - megaloblasts etiology:
-megaloblastic anemias result from conditions in which DNA synthesis in erythropoiesis is abnormal, for example due to
-vitamin B 12 deficiency
-folic acid deficiency
Both play role as cofactors in the synthesis of DNA
intrinsic factor= produced by mucosal cells in the stomach- intrinsic factor enables B12 vitamin to be absorbed properly from the food
-vitamin B12 is present in high concentrations in the liver and meat of animals, but is absent of plants- dietary B12 vitamin deficiency is rare, except of strict vegeterians
-folic acid is present in most vegetables- dietary deficiency is more common- in many states of malnutrition
4
there are two forms of megaloblastic anemias a) anemia due to B12 deficiency b) anemia due to folic acid deficiency a) Anemia due to B12 deficiency - has several possible causes, including so-called
A) PERNICIOUS ANEMIA
-is a form of megaloblastic anemia due to B12 deficiency
-it is common in western Europe and in the U.S. but is rather rare in Asia and Africa
-PA - occurs mostly after the age of 50
-slightly more often in males pathogenesis
-is an autoimmune disease caused by immunologic destruction of the gastric mucosa microscopically: - the mucosa of the body and the fundus of the stomach is characterized by diffuse lymphocytic infiltration and progressive atrophy of glands with a loss of parietal cells- chronic atrophic gastritis
- chronic gastritis is associated with a failure of secretion of acid
(achlorhydria) and intrinsic factor- leads to drastic reduction of B12 vitamin absorbtion
-three types of antibodies have been demonstrated in PA
-about 75% of patients - antibody that blocks vitamin B12 binding to intrinsic factor = blocking antibody
-about 50% of patients - antibody that binds with the intrinsic factor-B12 vitamin complex, thus interfering with binding of this complex to intestinal receptors
-about 90% of patients- antibodies against parietal cells the serum antibodies are of IgG type, in gastric juice-there are mostly IgA antibodies
-in some patients- no antibodies- proposed that the parietal cells may be destroyed even by T-cell mediated immunity pathologic and clinical features in PA:
-chronic atrophic gastritis –achlorhydria -failure of intrinsic factor secretion -leads to B12 vitamin deficiency
-megaloblastic anemia
-neurologic abnormalities due to demyelination (pathogenesis not clear) very characteristic-subacute combined degeneration of the spinal cord
(demyelination of the posterior and lateral collumns of the cord )
-leads to loss of position and vibration sense to loss of tendons reflexes
-peripheral neuropathy due to demyelination of nerves treatment
5
-replacement therapy by injected B12 vitamin- anemia improves rapidly and completely, neurologic symptoms recover slowly and incompletely patients with PA- are in greater risk of gastric ca
-precancerous epithelial dysplasia in chronic atrophic gastritis- can be detected by gastric mucosa biopsy other causes of B12 vitamin deficiency except of PA include:
- dietary habits- only in strict vegetarians
-after total gastrectomy
-patients with Crohn disease- chronic idiopathic inflammatory large bowel disease
-surgical removal of terminal ileum, -by-pass of terminal ileum - B12 vitamin complexed to intrinsic factor is normally absorbed in terminal ileum
-severe bacterial infection of large intestine
B) ANEMIAS DUE TO FOLIC ACID DEFICIENCY
-has several underlying causes including
-inadequate intake in food- in chronic alcoholism, in severe malnutritions
-due to failure of absorbtion- in malabsorbtive syndromes, such as tropical sprue, coelic disease
-this anemia occurs in the conditions of increased demand, such as in pregnancy, in malignant tumors- due to increased synthesis of DNA in malignant cells
-anticancer drugs- such as methotrexate - have antagonistic effect to folic acid-
Pathologic findings in all megaloblastic anemias:
RBCs changes- most apparent- erythropoiesis changes from normoblastic to megaloblastic megaloblast - larger than normoblast, shows delayed maturation but normal cytoplasmic hemoglobinization, primitive nucleus and fully hemoglobinized cytoplasm
changes in BM- accumulation of erythrocyte precursor cells - BM is hypercellular- resembles acute blastic leukemia
changes in peripheral blood- shows macrocytosis- large RBCs with anisocytosis-marked variation in size poikilocytosis-marked variation in shape
changes in neutrophils- neutrophil precursors in the BM show enlargment- giant metamyelocytes are characteristic in peripheral blood- hypersegmented nuclei in lekocytes
6
changes in other cells- disorder in DNA synthesis affects predominantly those cells with high rate of cell turnover (intestinal epithelium- enlarged nuclei )
Clinical symptoms in megaloblastic anemias
-severe anemia
-macrocytosis and hypersegmented leukocytes in peripheral blood
-bone marrow examination reveals hypercellularity- megaloblastic erythroid hyperplasia
4./ ANEMIAS DUE TO DEFECTIVE HEMOGLOBIN SYNTHESIS includes three major clinicopathologic entities a) iron deficiency anemia b) anemia in chronic diseases c) sideroblastic anemia
A) IRON DEFICIENCY ANEMIA
-is the most common type of anemia worldwide normal iron balance- is regulated mainly by changes in an intestinal absorbtion of iron -to substitute normal blood loss from the body- for example by secretion, menstrual bleeding, etc.
Causes of iron deficiencies
-iron deficiencies due to dietary deficiency- most common in underdeveloped countries
-increased demand of iron- in growth phase in early infancy or in adolescence in pregnancy and lactation
-malabsorbtion of iron- in severe generalized states, such as coelic disease, tropical sprue, etc
-after total gastrectomy- gastric acid is necessary for complete absorbtion of iron
-chronic blood loss- major cause of iron deficiency anemia results either from
-occult GIT blood loss due to hookworm infection- small parasitic intestinal worm (ancylostomiasis )= disease characterized by anemia, GIT pains, weakness -(larvae enter the body through the skin)
-occult GIT bleeding due to chronic ulcers, cancers, hemorrhoids, esophageal varices,
Pathologic findings:
-in iron deficiency- the first symptom- decreased serum level of ferritin- reflects the level of storage of iron -absence of iron in BM specimens when iron stores are exhausted- the serum iron level falls
7
-anemia is due to both a decresed amount of hemoglobin in individual RBC
(erythrocytes are poorly hemoglobinized) and decrease of total amount of RBCs erythrocytes are small- microcystosis- hypochromic microcytic anemia
-bone marrow changes- shows variable normoblastic hyperplasia, storage iron is absent
-epithelial changes- result in atrophy of many epithelial surfaces, such as mucous membranes of the mouth, tongue, stomach
Plummer-Vinson syndrom= iron deficiency anemia+ atrophic glossitis+ dysphagia
+koilonychia (concave fingernails) treatment:
-iron replacement therapy
-to find and correct the reason for anemia (ulcers, cancers)
B) ANEMIAS IN CHRONIC DISEASES
-in chronic renal failure- normochromic normocytic anemia due to failure of normal secretion of erythropoetin in the kidney
-BM shows mild erythroid hyperplasia
C) SIDEROBLASTIC ANEMIA
-is uncommon type of anemia characterized by the presence in the BM of increased numbers of sideroblasts= erythroid precursors with iron in their cytoplasm
-it has a variety of possible causes both primary and secondary- primary - may be inherited or acquired (of unknown causes) secondary- in chronic alcoholism, due to drugs, such as chloramphenicol, poisoning- chronic lead poisoning
Pathologic findings:
-SA is characterized by hypochromic microcytic or dimorphic anemia, it means that in the PB- mixture of normal erythrocytes, microcytes and macrocytic erythrocytes serum iron level increased
-BM shows increased amounts of storage iron, increased number of sideroblasts ring sideroblasts= normoblast in which the amount of Fe is greatly increased -
Fe granules surround the whole nucleus pathogenesis: defect in incorporation of iron into the hemoglobin molecule
II. ANEMIA OF BLOOD LOSS
acute- acute hemorrhage results in a loss of whole blood leading to hypovolemia- equivalent amounts of RBCs, white cells and serum are lost- thus, main blood values are normal- at first stage
8
-within few hours- there is compensatory water retention (important compensatory mechanism for hypovolemia) -results in decrease of RBCs count, decrease of hemoglobin concentration, decrease of hematocrit in PB
-regeneration of erythrocytes- BM shows erythroid hyperplasia
anemia is temporary, body iron storage is replaced over the next few months -chronic- chronic bleeding is compensated by erythroid hyperplasia of BM until Fe stores are exhausted
-after this point- iron deficiency anemia develops
III. HEMOLYTIC ANEMIA- due to increased rate of destruction of RBCs
-group of diseases characterized by shortened survival of RBCs in PB
RBCs destruction occurs
-in RES - extravascular hemolysis-common
-in blood - intravascular hemolysis -rare
EXTRAVASCULAR HEMOLYSIS-is characterized by
1. hemolytic jaundice-increased production of unconjugated bilirubin because of increased breakdown of hemoglobin
-unconjugated bilirubin is complexed with plasma albumin - transported to the liver- taken up by liver cells-jaundice develops when the amount of unconjugated bilirubin delivered to the liver exceeds the capacity of the liver to conjugate
2. increased level of bilirubin in bile- the liver excretes increased amounts of conjugated bilirubin into the bile- bilirubin pigment stones formation in the gallbladder
3. increased amount of urobilinogen- in urine, in large intestine content
4. erythroid hyperplasia- BM shows hyperplasia- expansion of BM cavities- increased erythropoiesis results in release of reticulocytes into PB ( in severe cases even- normoblast may appear in PB)
5. hemosiderosis-degradation of hemoglobin from the destroyed RBCs- deposition of hemosiderin (spleen, liver)
INTRAVASCULAR HEMOLYSIS is characterized by
1. hemolytic jaundice- unconjugated bilirubinemia
2. erythroid hyperplasia- in chronic hemolysis only
3. hemoglobinemie free hemoglobin in plasma appear when hemolysis has exhausted the capacity of plasma haptoglobin to bind hemoblobin
4. hemoglobinurie -hemoglobin molecule may pass the glomuruli
CLASSIFICATION OF HEMOLYTIC ANEMIAS
1) intrinsic defects of erythrocytes (intracorpuscular anemia) a- hereditary-
9
1- RBC membrane disorders or cytoskeleton disorders
-spherocytosis
-eliptocytosis
2- RBC enzyme deficiency
-G-6-PD (glucose-6-phosphatase-dehydrogenase deficiency)
3- hemoglobinopathies- hemoglobin synthesis disorders
-thalassemias
-sickle cell anemia
b-acquired - PNH
2) hemolysis due to extrinsic factors (extracorpuscular anemias)
immune anemia
other causes ad 1) intrinsic defects of erythrocytes ad 1a) hereditary:
HEREDITARY SPHEROCYTOSIS
-is congenital autosomal dominant disease with variable penetrance
-patients may present with severe hemolysis in childhood or mild hemolysis in adult age
-is characterized by change in shape of RBCs from the normal biconcave shape to a spherical shape- pathogenesis: proteins of red cell membrane are defective in structure or reduced in amount- results in reduction of cell surface- causes that RBCs assume a spheroidal shape
-spherical RBCs are more fragile- increased fragility - spherocytes are more susceptible to lysis- s. show autohemolysis- when incubated at 37 C for 24-48h
-life span of spherocytes is shortened- destruction occurs in the spleen clinical features:
-patients present with anemia and jaundice
-splenic enlargement- histologicaly splenic cords of Billroth are markedly congested and exhibit prominent erythrophagocytosis
-peripheral blood- shows spherical microcytes
-bone marrow shows normoblastic hyperplasia
-aplastic crisis may occur rapidly-associated with acute infection treatment: splenectomy- removal of the site of maximum erythrocyte destruction ad 2 ) GLUCOSE-6-PHOSPHATASE-DEHYDROGENASE DEFICIENCY (G6PD)
-most common erythrocyte enzyme deficiency- RBCs are more vulnerable to oxidants
-it is a X-linkedinherited anomaly-full expression of deficiency occurs in males hemolytic attacks trend to affect older RBCs clinically:
1 0
-most patients are asymptomatic, but acute intravascular hemolysis may occur due to exposure of oxidant drugs, such as sulfonamide, nitrofurantoin, etc.
-rarely patients develop mild chronic hemolytic anemia ad 3 ) hemoglobinopathies the most common Hb-pathies are:
thalassemias
sickle cell disease
1.THALASSEMIAS
-is a heterogenous group of congenital disorders, characterized by a lack or decreased rate of synthesis of either normal alfa- or the beta chains of hemoglobin A thalasemias are more common in Mediterranean , in Africa and Southeast Asia
beta-thalassemia- decrease of synthesis of beta-globin- free alfa chains form highly unstable aggregates - cell membrane damage- destruction of RBC precursors
alfa-thalassemia- inbalance in synthesis of alfa chain morphology:
-in peripheral blood- microcytic anemia with marked anisocytosis
-in bone marrow- ineffective erythropoeisis - due to destruction of RBC precursors in BM- erythroid hyperplasia
-in spleen- hemolysis of abnormal RBCs- activation- splenomegaly
-positive iron balance due to increased absorbtion of iron in the intestine and due to transfusions- lead to severe iron overload- secondary hemochromatosis- myocardial or liver failure
2.- SICKLE CELL DISEASE
-is a hereditary hemoglobinopathy caused by a single point mutation of the globin gene- that causes a replacement of aminoacids, of normal Hb glutamic acid for valine in beta chain -results in formation of hemoglobin-S morphology:
-occurrence of so-called sickle cells- elongated erythrocytes of pathologic shape, under decreased oxygen supply, pathologic erythrocytes bocome more elongated- called tactoids
-sickled cells are more susceptible to damage
-initially sickling of erythrocytes can be reversed to normal shape- when oxygenation improves
-later, however, the spindle shape of RBCs becomes permanent
-results in increased phagocytosis and destruction of the RBCs in the spleen clinically: onset in early childhood- death in young adult age
1 1
-patients present with chronic extravascular hemolysis and severe anemia- chronic hemolytic state- sickle cells have rigid membranes- prone to sequestration
-growth retardation
-mild hemolytic jaundice
-there is also tendency to microvascular occlusions- because sickle cells have a propensity to adhere to capillary endothelium
BM shows marked hyperplasia- compensatory normoblastic complications:
aplastic crisis- may cause sudden failure of hematopoiesis- may be due to acute infection, drugs, other causes
hemosiderosis- due to multiple blood transfusions and because of stimulation of absorbtion of iron in the intestine
vaso-occlusive crisis- is due to filling the microcirculation by aggregates of sickle cells- cause multiple small infarctions-painful episodes of ischemic necroses fever, ischemic pains-heart, skeletal muscels, bones-aseptic bone necrosis
splenic changes- are characteristic- enlargment, and due to repeated infarctions- multiple small scars with heavy hemosiderin deposition- called „autosplenectomy„- thus the patients have higher propensity to infections ad 1b)- acquired:
PAROXYSMAL NOCTURNAL HEMOGLOBINURIA (PNH)
-is a rare acquired disease of RBCs characterized by an increased sensitivity of RBC membrane to complement-results in chronic intravascular hemolysis
-complement activation may occur in vivo during sleep-because of decreased pHdue to slower respiration- results in paroxysmal nocturnal Hb-uria
-patients are young, anemia may be severe pathogenetically related to aplastic anemia-PNH represents a clone of abnormal erythrocytes developing in hypoplastic BM
2) hemolysis due to extrinsic factors (extracorpuscular anemias)
1- anemias mediated by antibodies- both autoimmune and isoimmune
2- mechanical trauma to RBCs
3- infections
4-chemical injury of RBCs- like lead poisoning
5- sequestrations of RBCs- for example in hypersplenism ad1.- IMMUNE-MEDIATED HEMOLYTIC ANEMIAS
1 2
among immune-mediated anemias, there are two major pathogenic groups- autoimmune and isoimmune
AUTOIMMUNE HEMOLYTIC ANEMIAS
-group of diseases in which hemolysis occurs as a result of the presence of auto-antibodies againts blood group associated antigens
-idiopathic warm autoimmune hemolytic anemia-characterized by presence of IgG (active at 37 C) – pathogenesis of hemolysis in most cases involves opsonization of RBCs by active IgG antibodies and subsequent phagocytosis by splenic macrophages.
-the clinical severity of immunohemolytic anemia is variable- most patients have mild chronic anemia with moderate splenomegaly and often no treatment is required
-idiopathic cold autoimmune hemolytic anemia- characterized by presenc of IgM antibodies- which have enhanced activity at temperatures below 30 C- hemolysis occurs after fixation of complement on IgM coated RBCs (this interaction occurs best in the distal part of the body)- then RBCs coated by
IGM and complement are phagocytosed by macrophages and removed from circulation
-cold hemolytic anemia is usually mild, formation of cold antibodies may be associated with lymphoproliferative disorders, or may be idiopathic
-Raynaud syndrome may occur in these patients because agglutination of RBCs in capillaries of exposed parts of the body
ISOIMMUNE HEMOLYTIC ANEMIAS
-are those in which the RBCs are destroyed as a result of the activity of antibodies of another person, this may occur for exmaple:
1-in blood tranfusion of incompatible blood- hemolytic reaction is rapid
-if severe - intravascular hemolysis results in hemoglobinemia such patient develop rapid shock- risk of death
2-hemolytic disease in newborn -due to Rh incompatibility
Hemolytic disease of newborn (erythroblastosis fetalis):
Is antibody-induced hemolytic disease in newborn that is caused by blood group incompatibility between mother and fetus.- this occurs if the fetus inherits red cell antigenic determinants from the father that are foreign to mother- particularly inmportant in this respect is Rh blood antigen
-although the incidence of hemolytic disease due to Rh incompatibility has decreased- this is because of successful prophylaxis
1 3
pathogenesis: pathogenetic basis of fetal erythroblastosis is immunization of the mother by blood group antigens onfetal RBCs and the free passage of mother antibodies to the fetus by placenta
-fetal red cells may reach mother circulation during last trimester of pregnancy or during child birth, the mother thus becomes sensitized, immuniziation depends on the dose of antigen- thus hemolatic disease develops only if the mother experienced sever placental bleeding
-hemolytic disease is thus uncommon in first pregnancy, the risk is increased in subsequent pregnancies
-currently all Rh-negative mothers are administred anti-D globulin soon after delivery of RH-positive baby. This globulin is linked with antigenic epitopes of fetal RBCs thus preventing long lastin immunization of the mother clinical course of erythroblastosis fetalis- vary from fetal hydrops (infants may be stillborn, or die within first days of life) to mild degree of hemolytic anemia, and may recover completely
3-drug induced hemolysis- the mechanisms of hemolysis induced by drugs are variable
- drugs such as metyldopa induce anemia indistinguishabel from primary idiopthic form of hemolytic anemia, autoantibodies are directed against intrinsic red cell antigens- mechanism how methyldopa induces formation of antibodies is not clear
-drugs such as penicilin,cephalosporines – likely bind to RBC membrane, and these drug-membrane complexes become antigenic. ad 2.- mechanical trauma to RBCs
MICROANGIOPATHIC HEMOLYTIC ANEMIAS
-is caused by fragmentation of RBCs as they pass abnormal microcirculation, Red cells may become demaged in a variety of circumstances, such as
-in DIC- fibrin strands- fragmentation of RBCs- hemolysis result in
-1) hemolytic uremic syndrome
-2) thrombotic thrombocytopenic purpura
-in abnormal blood vessels- in malignant hypertension
-in giant capillary hemangioma and malignant blood vessel tumors
-in prosthetic valves- causes trama to RBC, ad 3.- HEMOLYSIS CAUSED BY INFECTION
-several possible mechanisms may be involved including
-development of autoimmune hemolysis (infective mononucleosis, mycoplasma infections)
-production of hemolytic toxins (clostridia, streptoccoci)
1 4
-direct infection of RBCs- malaria -in malaria- red blood cell lysis occurs in attacks when a release of merozoites from infected RBCs occurs (Plasmodium vivax) fever, splenomegaly, diagnosis from blood smears (plasmodium may be visible)
POLYCYTHEMIA
= increased concentration of RBCs
- may be- relative and absolute
Relative: - due to decreased plasma volume dehydratation, such as due to loss of water in chronic long-lasting vomiting
Absolute:
- primary= increased number of RBCs due to intrinsic abnormal proliferation of stem cells =polycythemia vera this disorder is closely related to myeloproliferative syndromes
- secondary= increase in RBS mass in response to an increased stimulation caused by an increased level of erythropoetin
-may be appropriate- compensatory
- in lung diseases
- in people living for longer period in high-altitude
- in cyanotic heart diseases or inappropritate- pathological
- for example in erythropoetin secreting tumors, such as renal cell carcinoma
Grawitz,
- in hepatocellular carcinoma
- in cerebellar hemangioma
CHRONIC MYELOPROLIFERATIVE DISEASES
= group of disorders that result from clonal neoplastic proliferations of multipotent stem cells which may differentiate along one or more pathways
-predominant erythroid differentiation- polycythemia vera
-predominant paltelet differentiation- essential thrombocythemia
-predominant myeloid differentiation- CML multilineage differentiation is associated with marked marrow fibrosisosteomyelofibrosis (= myeloid metaplasia with myelofibrosis)
POLYCYTHEMIA VERA
= myeloproliferative disease characterized by excessive proliferation of erythroid, myeloid and and megakaryocytic elements derived of the multipotent stem cell and is associted with absolute increase in red blood cell mass because erythroid proliferation dominates pathogenesis:
1 5
- PV is associated with lower than normal levels of erythropoetin (in contrast to secondary polycythemias)
- absolute increase in number of stem cells, which are extremely sensitive to small amounts of erythropoetin
- PV is characterized by erytrocytosis=increased number of RBCs in PB morphology:
-RBCs appear normal
- bone marrow is markedly hypercellular with hyperplasia of all three components of active hematopoietic marrow, but predominantly of erythroid elements
-diffuse increase of reticulin meshwork in BM
-with progression of the disease-BM becomes more fibrotic= myelofibrosis or
BM may be replaced by blasts= leukemic transformation clinical features:
-occurs in middle-aged patients (4.-6. dec-)
-RBCs count more than 6-10 mil per mm3
-hematocrit more then 60 per cent (high viscosity of PB)
-total leukocyte and thrombocyte counts also increased major clinical features -result from an increased blood volume- increased viscosity-causes vascular stasis- thrombocytic tendency and hemorrhagic diathesis patients are plethoric= and slightly cyanotic (due to stasis and low oxygenation of slowly circulating blood)
-liver and spleen enlarged
-possible splenic and renal infarctions-pain
-headaches, GIT symptoms, hematoemesis and melena are common
-high cell turnover may cause hyperurikemia and symptoms of gout (5-10% of patients)
prognosis: about 30% of patients- die from thrombotic complications (brain stroke or IM) some may have hemorrhagic complications (GIT) in some the disease may develop to leukemia or OMF
OSTEOMYELOFIBROSIS (=MYELOID METAPLASIA WITH MYELO-
FIBROSIS)
=chronic MP disorder which is characterized by fibrotic obliteration of BM and metaplastic extramedullary hematopiesis principally in the spleen
-no identifiable cause- thus called „idiopathic (agnogenic) myeloid metaplasia„ morphology:
1 6
-PB variable- moderate to severe normochronic normocytic anemia (with poikilocytosis and increased number of normoblasts) white cells may be reduced or increased with a shift to left and few immature forms platalet count first normal, but with the progression of the disease- very apparent thrombocytopenia
-BM- shows diffuse fibrosis and obliteration of BM cavities, first by hypercellular ineffective hematopoiesis (composed of abnormal megakaryocytes and hyperplastic myeloid elements), later by fibrosis- in this phase the BM is hypocellular spleen-markedly enlarged- diffuse extramedullary hematopeiesis-trilineage proliferation liver-moderately enlarged-foci of EMH pathogenesis:
-unknown, proliferation begins in BM with subsequent proliferation in the spleen and liver fibrosis of BM is due to increased production of growth factor (PDGF-produced by the expanded pool of platelets and megakaryocytes) prognosis:
-most patients survive for years- sometimes transfusions are needed
-propensity to intercurrent infections thrombotic complications are rare, more common -bleeding disorders due to platelet abnormalities
-about 10% of patients reveal transformation to AL
LEUKOPENIA
=disorder of white cells characterized by decreased numbers of any one of the specific types of leukocytes, but most often the term implies decreased neutrophils
=neutropenia=AGRANULOCYTOSIS
=total white cell count is reduced to about 1 thousand and less per mm3 there is shortening of life-span of leukocytes (6-7 h) agranulocytosis develops rapidly causes:
1) inadequate or ineffective granulopoiesis
-due to suppression of stem cells ( aplastic anemias, variety of leukemias, lymphomas)
-due to suppression of granulocytic precursors ( for example after exposure to some drugs, toxins, etc)
-due to BM involvement for example in megaloblastic anemias
2) accelerated removal or destruction of leukocytes
1 7
-due to sequestration in the spleen - in hypersplenism-
-due to immunologically mediated injury to leukocytes- idiopathic or produced by drugs
-most significant leukopenias develop due to drugs:
-dose-related (predictable) BM suppression- caused by alkylating agents and antimetabolites used in cancer treatment
-idiosyncratic- (unpredictable), such as caused by aminopyrine, chloramphenicol, chlorpromazine, thiouracil, etc morphology:
-BM shows hypercellularity in leukopenias caused by increased destruction or in ineffective granulopoiesis, such as megaloblastic anemia
-or hypocellularity - when agranulocytosis is caused by agents toxic to precursors clinical course:
-fever, chills, weakness, fatique
-infections constitute the major problem (ulcerating necrotizing lesions of the gingiva, floor of mouth, buccal mucosa,etc) prognosis:
-difficult to predict better survival than before- thanks to supportive measures, such as neutrophil transfusions, antibiotics, steroids
1 8
NEOPLASMS OF HEMATOPOIETIC CELLS: LEUKEMIAS,
CLASSIFICATION FAB, MYELODYSPLASTIC SYNDROMES.
LEUKEMIAS
-malignant neoplastic proliferations of hematopoietic cells
-are characterized by diffuse replacement of the bone marrow by neoplastic cells
-majority of leukemias arise in BM, but in some types of leukemia bone marrow origin has not been proven as a site of origin- such as hairy cell leukemia
(spleen) and some CLL (lymph nodes)
-in most cases- neoplastic cells are also present in increased numbers in the peripheral blood
-leukemias are clonal processes that can develop de novo -primary leukemias or after bone marrow injury (after chemotherapy, radiation) or in myelodystplastic syndromes- secondary leukemias
Neoplasms of hematopoietic system differ from neoplasms of the other tissues in the following ways:
-hematopoietic tumors are often widespread at presentation- due to capacity of these cells to circulate by PB
-hematopoietic tumors do not usually form macroscopically apparent neoplastic masses, more likely they appear in a form of diffuse enlargement of the affected organ ( liver, spleen)
-benign proliferation of hematopoietic cells are common but benign hematopoietic tumors probably do not exist at all- in contrast to the frequency of benign tumors of other tissues
-hematopoietic tumors often change in appearance and clinical behavior during the course of illness due to changes in cell cycle or differentiation status
- for example CML arising from the pluripotent stem hematopoietic cell is typically composed of granulocytic cells in various stages of maturation, may transform into leukemia composed predominantly of immature myeloblasts= blast crisis
Leukemias are classified in several ways :
1) according to onset and clinical course
1. Acute - sudden onset - rapidly progressive course leading to death within several months if untreated usually characterized by primitive cells - called blasts (poorly differentiated )
2. Chronic - have an insidious onset and a slow clinical course the patients usually survive several years even if untreated
1 9
usually characterized by more mature neoplastic cells
2) according to the peripheral blood picture
1. leukemic - characterized by elevation of the white blood cell count in the PB and by the presence of neoplastic cells in the PB most common form
2. subleukemic - total white cell count is normal or lower, but leukemic
(neoplastic) cells are present in PB
3. aleukemic - total white cell count is normal or lower and no recognizable leukemic cells are to be found in PB
2. and 3. are very rare
3) according to cell type classification according to cell type is the most important of all and it becomes more complex as new criteria evolve for cell recognition there are two major groups of leukemias- FAB classification system
A) lymphocytic leukemias (FAB L1-L3)
B) myeloid leukemias- (FAB M1-M7)
-myeloid
-monocytic
-erythroleukemia these are very rare
-megakaryocytic leukemia
-plasma cell leukemia
-eosinophilic leukemia
MORPHOLOGIC FEATURES COMMON TO ALL LEUKEMIAS
1) Changes directly caused by leukemic infiltration leukemic cells may infiltrate any tissue or organ, but most striking changes are seen in: bone marrow-
-red-brown or gray-white color because of complete replacement of fatty BM by active neoplastic proliferation
-leukemic infiltrates may even erode cancellous or cortical bone spleen-
-massive splenomegaly is characteristic of CML (even more than 5-10kgs)- the enlarged spleen may virtually fill the abdominal cavity
-in CLL -enlargment of the spleen is less striking (2-3 kgs)
-acute leukemias produce only moderate splenomegaly (500-1000gs)
-enlarged spleens show higher tendency for infarctions (in CML) histologically:
-focal leukemic infiltrates with preserved normal structure
-in more progressive stages- diffuse massive involvement with total efacement of underlying architecture (replaced by homogenous infiltration by leukemic cells)
2 0
lymph nodes-
-enlargment is characteristic of some types of lymphocytic leukemias nevertheless, some infiltration of lymph nodes may be found in all types of leukemias histologically:
-obliteration of underlying architecture leukemic cells may invade capsule or sinuses (CML) or lymph nodes may be diffusely infiltrated (CLL)- picture identical to infiltration by malignant lymphoma liver-
-enlargment of the liver is somewhat more prominent in lymphocytic than in myeloid leukemias
-leukemic infiltrates are characteristically confined to portal areas in lymphocytic leukemias, whereas in myeloid leukemias, the infiltrates are illdefined (present within sinusoids and portal tracts) other tissues- any organ may be infiltrated in leukemias- leukemic infiltrates are frequently seen in the kidneys - small perivascular foci- later large progressively diffuse infiltrates
-infiltration of CNS- of particular importance in ALL because of protective effect of the blood-brain barrier, the leukemic cells may survive in the CNS even the chemotherapy and initiate a relapse- prophylactic radiation or intrathecal chemotherapy is administrated
2) Secondary changes due to inhibition of normal hematopoiesis bleeding diathesis-
-caused by thrombocytopenia- is the most striking clinical feature common to all types of leukemias
-hemorrhages may occur at any site, but the most common sites are:
-serosal linings of the bodies cavities
-serosal coverings of the viscera, particularly the lungs and the heart
(subpleural hemorrhages, subpericardial ecchymoses )
-mucosal hemorrhages- most common- into the gingiva, urinary tract
-intraparenchymatous hemorrhages- most important- the brain
DIC- common in M3- acute promyelocytic leukemia- may lead to bleeding disorders infections- a prominent feature- particularly common in oral cavity, lungs, skin, kidney may be caused by fungi, opportunistic infective agents
2 1
1) ACUTE LYMPHOBLASTIC LEUKEMIA (ALL)= malignant neoplasm composed of immature blastic elements with lymphoid differentiation derived of bone marrow stem cell and B- or T-lymphocyte precursors
-primarily it is a disease of small children and young adults
-it constitutes about 80% of acute childhood leukemias the age peak of about 4 years of age morphology:
-the nuclei of neoplastic blasts have coarse and clumped chromatin, one or two distinctive nucleoli
-in contrast to AML, the ALL blasts do not contain granules in the cytoplasm there are three morphologic subtypes of ALL according to FAB classification :
Classification system was developed by International group of hematologists and hematopathologists - is known as French-American-British
(FAB) classification
-ALL-L1
-ALL-L2
-ALL-L3 immunophenotype:
-about 80% of ALLs are derived of lymphocytes of B-type and in majority of patients - the blasts reveal pre-B or early B-lymphocytic differentiation
-only about 2 % of cases of ALL are of mature B-cell immunophenotype
-about 15% of ALL cases -blasts reveal T-cell markers
-less than 5 % of ALL cases- are unclassified ( nor B neither T-cell markers) treatment and prognosis:
-with chemotherapy- more than 90% of children may achieve a complete remission and more than 60% are alive 5 years later
-adults and children with T-cell ALL or B-cell with involvement of lymph nodes (so called leukemic phase of Burkitt lymphoblastoma) - have poorer prognosis
2) CHRONIC LYMPHOCYTIC LEUKEMIA (CLL) = malignant lymphoid neoplasm composed of small mature lymphocytes often with extensive BM infiltration and peripheral blood involvement
-is characterized by proliferation of small mature lymphocytes
-is the most indolent type of all leukemias
-it accounts for about 25% of all cases of leukemias
-it occurs typically in persons over 50 years of age (age peak 65), males are affected twice as often as females clinical course:
-often asymptomatic in early stage of disease
2 2
- patients may present with nonspecific symptoms, such as loss of weight, anorexia, etc
-in 60% of patients- generalized lymphadenopathy- lymph nodes are enlarged and reveal histologically a diffuse involvement by small neoplastic lymphocytes
(total effacement of original structure)
-hepatosplenomegaly is common
-in all cases- there is absolute lymphocytosis- small mature -looking lymphocytes
-diffuse infiltration of bone marrow by the same neoplastic cells- normal hematopoiesis remains present until advanced stage of the disease - then reduction of active BM-hemopoesis prognosis:
-extremely variable-depends on clinical stage (how many lymphoid areas are involved- liver, spleen, lymph nodes)
-depends on a presence of hemolytic anemia and or thrombocytopenia
-about 10-15% of patients develop auto-antibodies againts RBCs or platelets
-blast crisis or transformation to acute leukemia is very rare
3) HAIRY CELL LEUKEMIA ( HCL)
-distinctive form of chronic B-cell leukemia
-characterized by fine hair-like projections on the surface of neoplastic cells
-occurs in older adults ( 50-60%)
-more common in males morphology:
-PB shows normal white cell count in majority of patients, only about 25% of patients with HCL show leukocytosis
- PB smears in most cases- presence of „hairy cells„- B-lymphocytederived neoplastic cells- contain tartrate-resistent acid phosphatase (TRAP)- diagnostic for HCL typical morphologic features:
- bone marrow - infiltration-diffuse- BM shows hypercellularity and diffuse fibrosis
-leukemic cells in bone marrow- medium-sized, with ovoid nuclei, fine chromatin pattern and inconspicuous nucleoli, with abundant cytoplasm, cell membrane shows hairy processes
-massive splenomegaly - diffuse infiltration
-pancytopenia - in over 50% of patients (due to BM infiltration and splenic sequestration prognosis:
- prognosis is poor- median survival is 4 years
2 3
- splenectomy is of benefit in most patients, but results of surgical removal of the spleen are unpredictable
-disease responds poorly to chemotherapy (interferon helps in some patients) clinically: - patients usually present with anemia, neutropenia and massive splenomegaly
MYELOID LEUKEMIAS:
1) ACUTE MYELOID LEUKEMIA (AML) = is BM-derived neoplasm composed of myeloblasts and cells differentiating in granulocytic direction
-cell of origin is a granulocytic precursor cell
AML- extremely heterogenous group of leukemias
- most commonly affects persons between 15 and 40 years of age
- AML is characterized by proliferation of neoplastic myeloblasts diagnosis of myeloblast:
- presence of so called Auer rods (crystalline cytoplasmic inclusions seen in PB smears) when blasts show some degree of maturation into promyelocytes- coarse azurophilic granules and activity of chloracetate-esterase and myeloperoxidase appear
Clinical course:
- most patients present with symptoms related to BM failure
-40% have bleeding diathesis
-30% have severe infections some reveal weight loss, fever, hepatosplenomegaly most important clinical feature- increase in white cell count with 20-30% of blasts
AML is classified according to its morphology according FAB classification system into 7 categories:
M1-M4 - AML - leukemia arises from a multipotent myeloid stem cell in bone marrow
M1= acute myeloid leukemia without differentiation
-represent about 20% of all AMLs
-myeloblasts predominate, only some myeloblasts show myeloperoxidase activity, few Auer rods
M2= AML with differentiation
-represent about 30% of AMLs- myeloblasts and promyelocytes predominate, MPOX positive, Auer rods present
M3= acute promyelocytic leukemia
-5% of AMLs- hypergranular promyelocytes predominate, MPOX positive,
Auer rods positive
2 4
M4= acute myelomonocytic leukemia
-30% of all AMLs- myelocytic and promyelocytic differentiation is evident, MPOX positive, nonspecific esterase positive
M5= acute monocytic leukemia
-arises from multipotent granulocyte-monocyte precursor,
-represent about 10% of all AMLs- promonocytes or undifferentiated blasts, MPOX negative, monocytic-macrophage markers are positive (CD68)
M6= acute erythroleukemia
-less than 5% of all AMLs- rare- bizarre multinucleated blasts- PAS positive, glycophorin and hemoglobin positive, myeloid blasts (MPOX positive) may be also present
M7= megakaryoblastc leukemia
-less than 5% of AMLs- pleomorphic undifferentiated blasts reactive with anti-platelet antibodies (platelet-glycoprotein) typical feature- myelofibrosis (increased BM reticulin) prognosis:
- about 60-80% of patients achieve complete remission after intensive chemotherapy -average duration of CR is 1 year
- but long term disease-free survival is likely in only 10-15% of patients
-overall prognosis is worse than that of ALL patients
2) CHRONIC MYELOID LEUKEMIA (CML) = BM-derived neoplasm composed of granulocytic cells in various stages of maturation
- cell of origin = pluripotent stem cell, that can differentiate into myeloid, lymphoid or myelomonocytic cell lines
-this leukemia primarily affects persons between the ages of 20 and 60
-CML accounts for about 15-20% of all leukemis morphology:
- markedly elevated leukocyte count in PB
- circulating cells in PB are predominantly neutrophilic granulocytes or metamyelocytes and small number of myeloblasts may be also found in PB
(usually less than 10%) typically: presence of Ph chromosome „Philadelphia„ (translocation t9,22) clinical course and prognosis:
-initial symptoms- weakness, weight loss, fatigue, anorexia
-splenomegaly (hepatomegaly)- left lower chest pain- evidence of splenic infarctions due to vascular occlusion by aggregates of granulocytes
-bleeding diathesis and anemia may be present
-remissions may be induced by chemotherapy- with busulfan, hydroxyurea or other oral chemotherapy
2 5
- most pateints experience clinical improvement but Ph chromosome is not eradicated by this therapy nor is the final progression of disease altered
-most patients are likely to develop acceleration of disease or blast crisis accelerated phase of CML:
-is characterized by increasing anemia, thrombocytopenia- followed by blast crisis- transformation to AML blast crisis:
-may develop abrubtly without previous accelerated phase
-is characterized by increased numbers of undifferentiated blasts in PB and organs median survival is 3-4 years
BM transplantation may be of value during the chronic phase of disease-after development of blast crisis- all forms of therapy become virtually ineffective factors associated with a poorer prognosis- presence of palpable splenomegaly at presentation and excess in bone marrow myeloblasts
Myelodysplastic syndromes (MDS)
= group of hematopoietic disorders that are characterized by ineffective and disordered maturation of stem cells in the BM
-patients with MDS have hypercellular BMs but peripheral cytopenias associated with morphologic abnormaliteis in one or all cell lines
-BM is partly or totally replaced by a clone of stem cells that retain the capacity to differentiate along all three pathways of maturation, such as into
RBCs, granulocytes, platelets, but in a manner that is both ineffective and disordered
-MDS do not meet all criteria of malignancy but the stem cell clone has a tendency to lose an ability to differentiate, thus there is tendency to transform to AML
Morphologic and pathologic features: bone marrow biopsy:
- BM is hypercellular- abnormalities in all three components of hematopoiesis
-disorder of bone marrow topography- that may appear as a reversal of the usual location of individual components of the BM normally: myeloiesis is located pertrabecularly and erythropoiesis in central areas of bone marrow cavities
-disorder in maturation-
-megaloblastic erythroid hyperplasia
-hypogranular myeloid precursors
-increased proportion of blasts in BM
2 6
-micromegakaryocytes
-unilobed and bilobed neutrophils
-stromal abnormalities- perivascular fibrosis and increased reticulin
- PB shows pancytopenia
Diagnosis of MDS is based on hypercellular BM with trilineage dysplasia and peripheral pancytopenia
Clinical features:
-patients are usually elderly - most cases in 6th and 7th decade of life -
MDS is rare in individuals under the age of 60 years in older patients- MDS is likely to occur without a prior history of chemotherapy or radiation
-in younger people- MDS more often develops after BM injury
(chemotherapy or radiation) = secondary MDS
-more common in males
Prognosis:
-the overall median survival is 1-2 years, but MDS are prognostically diverse group of disorders-
-one third of patients progress to frank AML progression to acute leukemia occurs in diverse ways
-some patients show gradual increase in numbers of the BM blasts with increasing pancytopenia- that is finally followed by marrow failure and transformation to AML
-others show abrupt transformation to AML still others may show stable clinical course for many years- the only clinical symptom may be „ refractory anemia„- anemia unresponsing to common therapy.
PLASMA CELL DISORDERS
=this group of disorders is characterized by the proliferation of a single clone of immunoglobulin-secreting plasma cells
-followed by associated increase in serum levels of single homogenous IgG or its fragments thus these disorders are referred to as monoclonal gammopathies
- the monoclonal IgG can be identified in the blood and/or urine of patients with monoclonal gammopathies - is called M component
-the M component is complete IgG or light chain-called Bence-Jones protein- because of its small size is excreted in the urine
Variety of clinico-pathologic entities can be differentiated among monoclonal gammapathies (MG):
-multiple myeloma
-solitary myeloma
-Waldenstroms macroglobulinemia
2 7
-heavy-chain disease
-MGUS- monoclonal gammopathy of undetermined significance
MULTIPLE MYELOMA= plasma cell myeloma= Kahler Disease
=multifocal neoplasm arising in the bone marrow, typically composed of aggregates of neoplastic mature or immature plasma cells
-most common type of MG
Pathologic features: bone marrow:
-15-90% of bone marrow may be even replaced by plasma cell infiltrates these infiltrates appear as multifocal destructive bone lesions
-diagnostic for MM is massive replacement of marrow by homogenous sheets of mature or immature plasma cells
-neoplastic cells show polymorphism, small but distinctive nucleoli,
-neoplastic plasma cells are monoclonal- have the same heavy and the same light chain of immunoglobulin-
-individual bone lesions appear as sharply circumscribed destructive defects- any bone may be involved- but most frequently vertebral columns, ribs, skull kidneys:
-renal involvement- called myeloma nephrosis- in 60-80% of patients is histologically characterized by
-interstitial infiltrates of abnormal plasma cells or chronic inflammatory cells in the kidney
-protein casts consisting of albumin, immunoglobulins in distal collecting tubules often surrounded by multinucleated giant cell
-metastatic calcifications due to hypercalcemia
-pyelonephritis systemic changes:
-systemic amyloidosis of AL type may be found in some patients other organs:
-usually low tendency to progress to extramedullary sites-plasma cell infiltrates may be encountered in spleen, liver, lungs, nerve trunks,lymph nodes, etc.
Clinical features and complications:
-more often in males
-the disease is seen most commonly after the age of 50 years- the peak age incidence of multiple myeloma is between 50 and 60 years of age
-clinical features stem from the effects of infiltration of organs, particularly the bones and the bone marrow by the neoplastic plasma cells and by the production of massive amounts of immunoglobulins
2 8
-bone infiltrations is manifested
-by bone pains- most common initial complaint
-abnormal skeletal radiographs- show lytic bone lesions and pathologic fractures
-hypercalcemia resulting from the bone destruction may give rise to neurologic complications- confusion, lethargy, weakness
-then weakness, fatique- due to anemia ( in 60% of patients)
-most patients present with M component in the serum and Bence-Jones protein in the urine, minority show BJ proteinuria alone without serum M component
-there are recurrent infections- resulting from severe suppression of normal IgGs
-excessive production and aggregation of myeloma protein may lead to the hyperviskosity syndrome
-renal insufficiency appears in up to 50% of patients- result s from multiple factors- but probably the most important one is excretion of light chains-that are believed to be toxic to epithelial tubular cells
-rarely plasma cells may be found in the peripheral blood- giving rise to plasma cell leukemia prognosis:
-depends on the stage of disease at diagnosis
-about 70% patients have good response to chemotherapy- return of normal hematologic parameters = remision
-but the median survival is 2-3 years patients with multiple bone lesions, increasing levels of M component and increased levels of bence-Jones protein-poorer prognosis death- most common causes include infections and renal failure
SOLITARY MYELOMA OF BONE
= solitary bone marrow neoplasm consisitin of neoplastic plasma cells distinctive from the MM
-constitutes about 5% of MG
-elevated levels of M protein in the blood or urine are found in about 25% of patients
-it is different from the MM in the absence of anemia or renal involvement
-the solitary bony lesions tend to occur in the same locations as MM and progress to MM in most cases
EXTRAMEDULLARY PLASMOCYTOMA
=is a solitary extraosseous lesion composed of neoplastic IgG-secreting cells
-frequently located in the lungs, in the upper respiratory tract (pharynx, paranasal cavities), rarely seen in oral cavity, stomach better prognosis than MM
2 9
- rarely disseminate, can be cured by local excision
WALDENSTROM MACROGLOBULINEMIA
-constitutes about 5% of MG and shares some features with MM and small lymphocytic malignant lymphoma - the M component is usually IgM type- macroglobulinemia morphologic features:
-diffuse infiltrates composed of plasma cells, plasmacytoid cells and lymhpocytes in the marrow- no bone erosions
-tumor cells may be also found in lymph nodes, spleen, liver clinical features:
-presents usually in older patients- most common between 6th and 7th decades
-nonspecific complaints, such as weakness, fatigability, weight loss
-approximately 50% of patients have lymphadenopathy, and hepatosplenomegaly
-macroglobulins greatly increase the viscosity of the blood-giving rise to so called hyperviscosity syndrome-that is characterized by:
-neurologic problems resulting from the sluggish blood flow- headaches, dizziness, deafness
-bleeding related to hyperviscosity due to dysfunction of platelets
-cryoglobulinemia-abnormal globulins may precipitate at low temperatures- producing for example Raynaud s syndrome
Prognosis : the average survival with chemotherapy is 2-5 years
HEAVY-CHAIN DISEASE
= very rare monoclonal gammopathy characterized by elevated levels in the blood or urine of a specific heavy chains. three variants - based on the chain involved
-gamma-chain-disease encountered most often in old patients is characterized by lymphadenopathy,anemia and hepatosplenomegaly
-alfa-chain-disease is most common type of heavy-chain disease, is a disorder of Ig-A-secreting cells is characterized by massive infiltrates in mucosa of the intestine and in abdominal lymph nodes composed of plasma cells, lymphocytes, histiocytes
-MU-chain disease is the rarest type, encountered in some patients with CLL present with hepatosplenomegaly
MONOCLONAL GAMMOPATHIES OF UNCERTAIN SIGNIFICANCE
3 0
-characterized by presence of M protein in the blood without any of the well-defined immunoglobulin-producing disease
-the patients have usually less than 5% of plasma cells in the bone marrow
-benign clinical course without any symptoms, but about 18% of patients develop a well-defined plasma cell dyscrasia later ( MM, macroglobulinemia, amyloidosis or lymphoma)
3 1
BLEEDING DISORDERS
- disorders characterized by an increase in tendency for bleeding, may be caused by
I.-increased fragility of blood vessels
II.-disorders of platelets
III.-defects in coagulation or by combination of all factors
I. DISORDERS DUE TO INCREASED VASCULAR FRAGILITY
-disorders in this group are relatively common, but usually do not cause serious bleeding
-most of them result in multiple petechiae or purpuric hemorrhages
-platelet count is normal possible conditions resulting in this type of bleeding diathesis include:
-infections- underlying mechanismes are vasculitis or DIC
-drug reactions-often mediated by immune complexes in the vessel walls with production of a acute vasculitis
Henoch-Schonlein purpura- a systemic hypersensitive reaction of unknown cause, characterized by abdominal pains, polyarthralgia, acute glomerulonephritis- associated with deposition of immune complexes
II.-DISORDERS OF PLATELETS
A) decrease in number of platelets -THROMBOCYTOPENIA
B) defective function of platelets
THROMBOCYTOPENIA
=decrease in platelet number ( normal count 150-300 thousands per mm3)- decrease to 10 thousands and lower levels causes bleeding tendency
-characterized principally by petechial bleeding- most often from small vessels of the skin and mucous membranes possible causes include:
-decreased production of platelets- occurs in generalized diseases of the BM, such as in aplastic anemia,disseminated cancer, leukemias
-decreased platelet survival-results from immunologically medited destruction of platelets- may follow drug administration, such as methyldopa or infections, for example AIDS
-there is compensatory megakaryocytic hyperplasia
-sequestration-may occur in the presence of splenomegaly -the syndrom is called hypersplenism
-splenectomy can cure
3 2
most common clinicopathological entities associated with thrombocytopenias are:
1.-IDIOPATHIC THROMBOCYTOPENIC PURPURA ( ITP)
-is associated with immunologically mediated destruction of platelets two forms are recognozed: acute ITP -is a self-limited disorder- most common in children after viral infections ( viral hepatitis, infectious mononucleosis, rubeolla, cytomegalovirus infections, etc. )
-platelet destruction is probably caused by antigen-antibodies complexes, directed against viruses, absorbed however on platelets chronic ITP -destruction of platelets results directly from the presence of anti-platelets antibodies
-in most patients the platelet-associted IgGs can be identified
-destruction of antibody-coated platelets occurs in the spleen-splenectomy is beneficial for more than 80% of patients clinical features of ITP:
-chronic ITP occurs most often in women, of medium age
-it may be associated with other autoimmune disorders, such as autoimmune hemolytic anemia,systemic lupus erythematodes,etc.
-most commonly slowly progressive course, with petechial chronic bleeding, such as nose bleed, but sometimes more serious complications like for example melena, hematuria or intracerebral hemorrhages may appear
2.- ISOIMMUNE THROMBOCYTOPENIA
-group of disorders that result from development of antibodies directed againts specific platelet antigens
-after transfusions
-neonatal thrombocytopenia- similar pathogenesis as in fetal erythroblastosis ( platelet-antigen-negative mother carrying antigen-positive baby develop antibodies againts platele-antigen that may cross the placfenta and cause thrombocytopenia in the newborn
3.- THROMBOCYTIC THROMBOCYTOPENIC PURPURA (TTP)
-rare disorder characterized by thrombocytopenia, microangiopathic hemolytic anemia and fever, neurologic symptoms and renal failure
-most clinical symptoms are due to formation of multiple hyaline microthrombi pathogenesis immunologic reaction is directed against endothelial cells, results in pathologic aggregation of platelets- formation of microthrombi- in contrast to
DIC, activation of clotting system is not primary clinically: more commonly in females- peak age about 40years of age treatment with corticosteroids
3 3
II-B. HEMORRAGIC DISORDERS RELATED TO DEFECTIVE PLATELET
FUNCTION
-characterized by prolonged bleeding time associated with normal platelet count may be congenital or acquired congenital -there may be defects in platelet adhesion ( congenital deficiency of platelet-membrane glycoprotein) or defects in platelet secreting activity-platelet anable to produce prostaglandins, etc. acquired- many possible conditions, of which two are of clinical importance aspirin is known to supress the synthesis of thromboxane which is necessary for platelet aggregation- used in supplementary treatment of IM bleeding in uremic patients may be partly due to defects in platelet function
III.- HEMORRHAGIC DIATHESIS DUE TO ABNORMALITIES IN CLOTTING
FACTORS
-bleeding seen in patients with abnormalities in clotting system in contrast to bleeding in platelet deficiencies is characterized by
-more often the bleeding is not spontaneous, more likely develops after injury or surgical procedure, but is usually serious, with prolonged bleeding and large hematomas and hemorrhages
-most common sites of bleeding is the GIT, urinary tract and particularly large joints and the soft tissues in their vicinity
-clotting abnormalities may be due both to hereditary disorders, such as in hemophilia and von Willebrand Disease- it was already mentioned before or may be acquired-most important clinical settings in this respect are the vitamin K deficiency, most often caused by liver failure and DIC- that produces a deficiency of multuple coagulation factors
DISSEMINATED INTRAVASCULAR COAGULATION ( DIC)
-is an acute, subacute or chronic thrombohemorrhagic disorder occurring as a secondary complication of a variety of diseases:
-is characterized by activation of the clotting system leading to formation of multiple microthrombi throughout the microcirculation- the consequence of which is consumption of platelets, fibrin and coagulative factors, secondarily leading to activation of fibrinolytic system thus, the DIC presents with
-signs and symptoms related to infarctions caused by microthrombi
-signs of hemorrhagic diathesis due to depletion of the elements necessary for hemostasis and even due to secondary activation of fibrinolytic processes pathogenesis:
3 4
there are two major mechanisms by which the DIC may be triggered:
1) release of tissue factors and thromboplastic substances into the circulation
-these substances may be derived from various sources, such as placenta in various obstetric complications, bacterial endotoxins, particularly in gramnegative sepsis, etc.
2) widespread injury to the endothelium -endothelial injury can initiate DIC by causing release of thromboplastic factor from endothelial cells, by promoting the platelet aggregation, by activating the intrinsic pathway of clotting cascade
-this endothelial injury may be produced by depositions of antigenantibodies complexes morphology:
-microthrombi with infarctions in multiple organs and hemorrhages clinically significant changes are encountered in : kidneys- thrombi in glomeruli and renal cortical microinfarcts lungs- microthrombi in alveolar capillaries- may cause clinically acute respiratory distress syndrome brain- microinfarcts and fresh hemorrhages placenta-widespread thrombi clinical features: about 50% of patients with DIC are abstetric patients with complications of pregnancy
- sepsis and major trauma- rest of patients other causes- rare major clinical manifestations of DIC:
-microangiopathic hemolytic anemia resulting from microvascular occlusions
-respiratory symptoms including cyanosis, dyspnea
-neurologic symptoms including convulsions
-oliguria and renal failure
-circulatory failure and shock the prognosis and treatment is highly variable and depends on the underlying disorder- administration of either aggressive anticoagulants, such as heparin and antithrombin
-or coagulants usually in the form of fresh-frozen plasma
3 5