Jizhong Zhou, PhD
advertisement

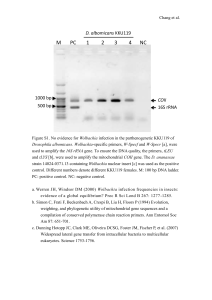
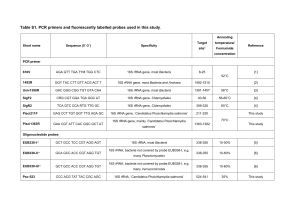
April 12, 2010 Jizhong Zhou, PhD Editor, Applied and Environmental Microbiology (AEM) Re: Diversity of 16S rRNA genes within individual prokaryotic genomes (AEM02953-09 Version 1) Dear Dr. Zhou, Thank you for reviewing our manuscript, “Diversity of 16S rRNA genes within individual prokaryotic genomes” (AEM02953-09 Version 1). We appreciate the opportunity to have the updated manuscript considered for re-review. The reviewers’ comments have been very helpful in improving the manuscript’s quality and content. Please find below an item by item disposition of each of the reviewers’ suggestions, followed by our responses. Sincerely, Zhiheng Pei, MD, PhD REVIEWER 1: 1. Several times the manuscript give 3% difference in 16S rRNA as the species boundary "by general definition" (p 17 li 19), citing the 1994 work of Stackebrandt and Goebel. The authors are apparently unaware that Stackebrandt later revised his estimate to between 1 and 1.5 percent (Stackebrandt & Ebers. Microbiology Today, Nov 2006 pp. 153-155.) We would like to thank the reviewer for providing us with the update on the operational definition of species using 16S rRNA gene sequences. Stackebrandt & Ebers suggested using 1-1.3% (not 1-1.5%) to replace the old value of 3% as the species boundary. With the new values, intragenomic variation of 16S rRNA genes exceeds this boundary in 24 species (see updated table 2). We have incorporated the new definition into our manuscript. Please note that much of the changes in the revised manuscript are related to the update on this definition. 2. Normally, for phylogenetic analysis, only those positions that can be well-aligned between most sequences are included in the analysis. Hypervariable regions and 1 regions of variable length and secondary structure are usually "masked out". It would greatly improve the paper if, in addition to the overall percent difference, percent differences were calculated after using such a mask. The 1991 Lane mask would be a good choice here. (Lane,D.J. 1991. 16S/23S rRNA sequencing. In Stackebrandt,E. and Goodfellow,M. (eds), Nucleic Acid Techniques in Bacterial Systematics. John Wiley and Sons, New York, pp. 115–175.). We appreciate the reviewer for this thoughtful suggestion. As suggested, we aligned 16S rRNA genes from the 24 highly diversified species listed on the updated Table 2 and masked the aligned sequences with Lane mask. Intragenomic differences were recalculated on the masked sequences and the results are added in Table 2. We have updated the Methods section with this new analysis. 3. Related to the above comment, if a high proportion of the changes occur in these hypervariable regions that are not normally used for phylogenetic inference, then the portions of the manuscript dealing with the effect on inter-organism comparisons will need to be revised. Of the 24 highly diversified species listed on Table 2, the effect of masking hypervariable positions was remarkable for 14 species, reducing the diversity from between 1.06% and 2.07% to <0.66% (Table 2). This level of diversity will not have a significant impact on phylogenetic inference. However, the variation after masking remained high for H. marismortui (4.86%) and T. tengcongensis (5.01%), Candidatus Protochlamydia amoebophila (1.53%), Carboxydothermus hydrogenoformans (1.24%), Deinococcus geothermalis (1.09%), and Geobacillus thermodenitrificans (1.09%). For B. afzelii, the two 16S rRNA genes are too diversified to be aligned using available algorithms (Our original alignment of full sequences of these two genes was done manually). Such diversified 16S rRNA genes will be troublesome not only for threshold-based taxonomic assignment using full length sequences but also for phylogenetic inference using masked sequences because 16S rRNA genes from within the same genome are not monophyletic in these species. We have updated the Results section with these new data. 4. A more complete position-by-position comparison of the intra-genomic vs inter-genomic rates of changes should be presented. In the current manuscript this is done for only a single organism (fig 3). We presented this type of analysis with two examples, Thermoanaerobacter tengcongensis (Figure 1) and Borrelia afzelii (Figure 4). These examples were selected to demonstrate important conclusions from this study. T. tengcongensis was used to demonstrate the power of ribosomal constraint on at the secondary structure level, while B. afzelii was used to show loss of ribosomal constraint in a pseudogene. Although more figures like these can be included in the manuscript, they are too complex as Reviewer 3 pointed out (See question 8 from Reviewer 3). To balance between the suggestions by Reviewers 1 and 3, we would like to keep Figs. 1 and 3 unchanged to preserve the details asked by Reviewer 1 but will not add more figures like these as it would exaggerate the concern raised by Reviewer 3. 2 5. p 12 li 3 should be "Table 3",not "Table 1" Table 1 now has been changed to Table 3. 6. Figure 1. I can't distinguish the "large letters" mentioned in the legend. We have changed the “large letters” to “colored letters” in the legend. 7. Order of figures doesn't match first use in text? The figures have been reordered in the order of first appearance. REVIEWER 2: 1. Minimum-free energy folding on single sequences is widely known to perform poorly. Strongly recommend re-doing this part of the analysis using constraints on conserved regions/folds and-or using multiple-sequence folding approaches, which work much better (see e.g. Paul Gardner's reviews on this topic). We understand the reviewer’s concern with minimum-free energy folding on single sequences. The minimum-free energy folding approach we used effectively predicted ribosomal constraint at the 2º structural level for nearly all species without need for alternative folding strategies. We believe this success was due to the availability of consensus 16S rRNA models that were used to guide the folding. However, we encountered difficulty, as the reviewer predicted, when using the same approach to fold the whole 16S rRNA molecules for 5 species, S. woodyi, P. profundum, C. cellulolyticum, Desulfitobacterium hafniense, and Syntrophomonas wolfei (Table 3 and Fig. 3). The difficulty was caused by a high concentration of substitutions in certain regions of these 16S rRNA molecules, which prevented more detailed comparison. As the reviewer suggested, for 16S rrn genes that displayed high levels of regional diversity, the regions in question were folded using the KnetFold program (Bindewald 2006). This folding method creates secondary structures based on multiple sequences. The output from KnetFold was entered into jViz.RNA 2.0 in order to visualize the secondary structure (Wiese 2005). jViz.RNA 2.0 allows for the creation of complex secondary structures that may contain pseudoknots. The multiple sequence folding was verified using another program named Murlet (Kiryu 2007) (see update in Methods section). The results were included in Results and Discussion sections and Fig. 3. 2. What implications do the results have for chimera detection? If homogeneity within a genome is maintained by gene conversion, do the recombinants cause problems for chimera detection algorithms? As the reviewer suggested, we checked for chimeras in all 16S rRNA genes from species listed on Table 2, using Bellerophon (Huber 2004). No chimeras were detected. This outcome was somewhat expected as chimera detection relies on obvious breakpoints where two 3 phylogenetic distinct parent molecules are ligated. Such subtle recombinations would be below the typical sensitivity of chimera detection algorithms as commonly employed. 3. Would be useful to add to table 1 min, max, and standard deviation of # rRNA genes per genome. Could the authors comment on how copy number variation is likely to bias our 16S-based estimates of community composition, and whether these biases are likely to matter in practice? Table 1 has been updated with the min, max, and standard deviation of # rRNA genes per genome, as recommended. Since inter-quartile range and median were used to describe the data in the original manuscript, this change created an inconsistency between the tables and the manuscript. To fix this inconsistency, inter-quartile range and median were replaced with min, max, and standard deviation in the revised manuscript. It is well known that there is wide variation of copy numbers of 16S rRNA gene among various species (Lee 2009, Rastogi 2009). Currently, it is common practice to describe the composition of a microbial community using 16S gene composition rather than cell composition. It would be desirable to convert 16S gene composition to cell composition but for a large number of organisms in a complex microbiome, this conversion is not possible because of the lack of knowledge about the copy numbers of 16S rRNA gene in their genomes. Let’s illustrate the difference using an artificial example in which a microbial community contains 100 bacterial cells, 90 cells from Borrelia turicatae and 10 cells from Brevibacillus brevis. Because there is one 16S rRNA gene per cell for B. turicatae and 15 16S rRNA genes per cell for Brevibacillus brevis, this community contains 240 16S rRNA genes, 90 from B. turicatae and 150 from Brevibacillus brevis. Consequently, this community is dominated by cells from B. turicatae (90/100) and by 16S rRNA genes from Brevibacillus brevis (150/240). Thus, 16S gene composition is an acceptable way to describe a microbial community with the understanding of the difference between the 16S gene composition and cell composition. This discussion now has been included in the revised manuscript. 4. Are there any changes in diversity correlated with differences in GC content? There is no correlation for most species except for the top three species with the highest diversity. Besides the two species, H. marismortui and T. tengcongensis, discussed in the original manuscript, the discussion has been updated with B. afzelii. Of the two 16S rRNA genes in B. afzelii, the pseudogene has a much lower GC content (38.1%) than the functional copy (46.5%). It appears that random mutations in the pseudogene have been bringing its GC content towards the baseline for the whole genome (28%). 5. How do the variability estimates in Fig. 1. compare with traditional estimates of variability from environmental sequencing projects? To our knowledge Thermoanaerobacter tengcongensis, as shown in Fig. 1, harbors the most diversified 16S genes among all known prokaryotic species except for Borrelia afzelii whose 4 high diversity is related to a pseudogene. This level of diversity is comparable to those found by using traditional PCR cloning technique in Haloarula marismortui (5%) and Thermobispora bispora (6.4%). 6. Does the availability of high-quality complete genome sequence allow the avoidance of low quality read problems that can artificially increase variability in estimates from single-pass environmental sequencing projects? No. The genome sequences are helpful but limited in this regard because the genome database only covers a very minor fraction of true variations of 16S rRNA genes in natural world. The database is too small to allow identifying or correcting sequence errors by cross reference to 16S rRNA genes in the database. REVIEWER 3: 1. P11L14 - Any evidence that this gene is expressed in B afzelii? Also, is there any evidence for horizontal gene transfer or is this merely the accumulation of deleterious mutations? This is an important consideration as intra-genomic variation is often used by the Ford Doolittles of the world to critique the use of 16S rRNA genes to infer organismal phylogeny? There has not been any experiment designed to examine the expression of rrnA of B. afzelii. It does not appear to be horizontally transferred into B afzelii from other species, as it is closer to rrnB of B. afzelii than to a species in any other genera. We have updated P11L14 with these sentences. 2. P14L16 - What is the evidence that genes have been lost? Do other genes in operon appear to be pseudogenes? We understand the reviewer’s concern on the word “lost” or complete deletion of 16S rRNA genes. Now, instead of implicating there was an event of lost or deletion, we simply describe the status of the involved rRNA operons as partial rRNA operon missing 16S rRNA gene. We updated P14 with the following information. Missing of a whole 16S rRNA gene in a rRNA operon, as evidenced by the presence of 23S or 5S rRNA genes but absence of 16S rRNA gene, was observed in rRNA operons in 95 species (Table S2.). This ranges from an absence of one 16S copy to an absence of eight copies in S. wolfei. The 23S or 5S rRNA genes in the partial rRNA operon appear functional because none of the genes exhibit excessive random mutations characteristic of a pseudogene. Interestingly, intragenomic diversity among 16S rRNA genes in 6 of the 95 species was borderline or slightly above the 1-1.3% threshold for separation of species (Table 2). These species include Shewanella sp. ANA-3 (1.09%), Escherichia coli (1.10%), Bacillus clausii (1.15%), Bifidobacterium adolescentis (1.30%), Shewanella baltica (1.36%), and Syntrophomonas wolfei (1.67%). As described before, the high diversity in S. wolfei was also associated with IVS in 16S rRNA genes. 5 3. P17L19 - I know the authors probably feel compelled to comment on the concept of an operational species definition. A larger point could be made that 16S rRNA genes probably evolve at different rates. In fact, this would be a very opportune time to look at how intra- and inter-genomic variation compares within and between species. We have updated the manuscript with the following discussion, as recommended by the reviewer. The definition for prokaryotic species is polyphasic in that it requires a distinct set of biological characteristics and corresponding DNA reassociation values greater than 70%. However, there is not a simple, universal definition. 16S rRNA genes have been used as a surrogate maker for operationally defining species. Initially, >3% difference between 16S rRNA genes from two organisms was required to claim the two organisms belong to two different species (Stackebrandt 1994). Later, the threshold was lowered to 1-1.3% (26). This operational definition is helpful in taxonomic classification using 16S rRNA genes, especially for studies of complex microbiomes using cultivation-independent techniques in which biological characteristics and DNA reassociation values can not be determined for individual bacterial cells/species. Nevertheless, it is critical to understand the limitations of the 16S rRNA-based operational definition for species. The main limitation is that 16S rRNA genes evolve at different rate but the operational species threshold (1-1.3%) is relatively rigid. As a consequence, closely related species that evolve slowly will be grouped as a single species by the operational definition such as, Streptococcus pseudopneumoniae and Streptococcus pneumoniae (Arbique 2004) that differ by only 5 bp between their 16S rRNA genes corresponding to only 0.03% difference. Another limitation is that 16S does not represent the entire genomic content that determines the biological characteristics for a species. It is by now quite evident that significant differences in genome composition may be present in bacterial species that are completely identical or that differ only slightly in 16S rRNA genes. For example, isolates of Vibrio splendidus exhibit up to 25% genotypic difference (Thompson 2005), and strains of E. coli may differ up to 40% in the number of genes in their genomes (Perna 2001, Kudva 2002). The three members in the Bacillus cereus group, B. cereus, B. anthracis, and B. thuringiensis can be classified as a single species by their nearly identical 16S rRNA genes but differ greatly by the number and type of genes they harbor due to the presence of large plasmids (Rasko 2005). In both E. coli and the B. cereus group, these differences confer various biological capabilities and pathogenicity. Intragenomic variation of 16S rRNA genes is another limit that can be encountered when classifying species that harbor 16S rRNA genes with diversity greater than threshold set by the operational definition (Table 2), which will lead overestimation of species diversity in a microbiome. Thus, it can be expected that community structures determined using the 16S-based operational species definition approximate but do not necessarily reflect the true community structures. 4. The authors comment that intra-genomic variation could confound taxonomic classifications. It would be interesting to see whether 16S rRNA gene from within the same genome are monophyletic. 6 Please see reply to question 3 from reviewer 1. 5. P8L6 - 19 Archaea + 408 Bacteria = 427 total - not 425 We sincerely thank the Reviewer for the careful review of our data. The error was due to our partial correction of a miscalculation. Actually, there are 425 species including 19 Archaea + 406 Bacteria. Finding this error promoted us to verify all numbers in the Tables. No additional errors were identified in Tables 1-3. However, two errors were found in Table S1 (568 prokaryotic species analyzed in this study). Table S1 included two genomes for Vibrio fischeri. Removal of the redundant Vibrio fischeri genome reduced the number of unique species in this study to 588 from 569. The number of 16S rRNA gene in Mycobacterium laprae was zero but should be one. Four species were removed from Table S2 (95 prokaryotic species with partial rRNA operons missing 16S rRNA genes) because the species either had no partial rRNA operons (Streptomyces coelicolor) or had partial rRNA operons missing 23S rRNA genes instead of missing 16S rRNA genes (Persephonella marina, Sulfurihydrogenibium azorense, Vibrio harveyi). 6. P9L3 - How are distances calculated when there are IVS's? If gaps were determined to be caused by intervening sequences (IVS) (inserts >10 bp), they were recorded and removed and sequences were realigned and distance recalculated. Please see P6L14 in the Method section for detail. 7. P10L13 - Possible to state the variable regions that these occur? P10L13 has been updated using variable regions, as recommended. 8. The tables and figures are brutal. Is there anyway to simplify these to highlight the important points? Please see reply to Question 4 of Reviewer 1. 9. Organismal names need to be italicized throughout. All organismal names have been italicized in the revised manuscript. 7