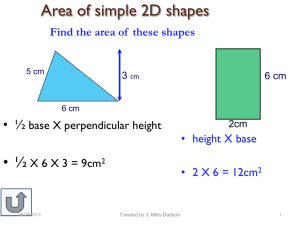
View/Open - Sacramento
advertisement

THE CREATION OF GLYCODENDRIMERS FOR THE INHIBITION OF HIV USING THE DISACCHARID MALTOSE AND TRIS 2-(AMINOETHYL)AMINE Rachel Ann Blackeye B.A., California State University, Sacramento, 2008 THESIS Submitted in partial satisfaction of the requirements of the degree of MASTER OF SCIENCE in CHEMISTRY (Biochemistry) at CALIFORNIA STATE UNIVERSITY, SACRAMENTO FALL 2011 THE CREATION OF GLYCODENDRIMERS FOR THE INHIBITION OF HIV USING THE DISACCHARIDE MALTOSE AND TRIS 2-(AMINOETHYL)AMINE A Thesis by Rachel Ann Blackeye Approved by: __________________________________________, Committee Chair Katherine Dawn McReynolds, Ph.D. __________________________________________, Committee Member Mary McCarthy-Hinz, Ph.D. __________________________________________, Committee Member Cynthia Kellen-Yuen, Ph.D ___________________________ Date ii Student: Rachel Ann Blackeye I certify that this student has met the requirements for format contained in the University format manual, and that this thesis is suitable for shelving in the Library and credits is to be awarded for the thesis. __________________________, Graduate Coordinator ___________________ Susan Crawford, Ph.D. Date Department of Chemistry iii Abstract of THE CREATION OF GLYCODENDRIMERS FOR THE INHIBITION OF HIV USING THE DISACCHARIDE MALTOSE AND TRIS 2-(AMINOETHYL)AMINE by Rachel Ann Blackeye The human immunodeficiency virus (HIV) affects people around the globe. According to the National Institutes of Health, around 33.3 million individuals worldwide are infected with HIV-1, and within this group more than 1 million people are found in the United States. There are therapeutic treatments used to fight HIV before it develops into AIDS (acquired immunodeficiency syndrome), but they are not without drawbacks. Viral resistance and harmful side effects are problems that have led to new research directed towards fighting this virus. Most of the drugs created thus far attack the virus from within the host cell. Scientists are now looking at targets on the viral surface that would prevent the virus from entering the cell. Dendrimers, which have been around since the mid 1980s, have the potential to block viral entry through the multivalent effect. Dendrimers are branched macromolecules that are globular in shape, and structurally well-defined with multivalent reactive terminal groups. As these molecules branch out from the core, new generations iv of compounds may be added to double the number of reactive ends. When the end groups on a dendrimer are carbohydrates, it is known as a glycodendrimer. There are two processes used to formulate glycodendrimers called the divergent and convergent methods. In the divergent method, dendrimers are synthesized from the core outwards. With the convergent technique, the outer portions are synthesized first and then added to the core. Dendrimers have been utilized in a variety of ways since their creation in 1984. In some studies, glycodendrimers have demonstrated anti-HIV properties. The goal in this research was to create two glycodendrimers using both the convergent and divergent processes. The hexavalent maltose amino-oxime glycodendrimer was synthesized (66% yield) using the convergent method and the trivalent maltose amino-amide glycodendrimer was created (3.4% yield) via the divergent method. A hexavalent amino core (16% yield) was synthesized as well, which will be used in later research to create other glycodendrimers. In the end, once these glycodendrimers are sulfated, they will be evaluated in a competitive gp120 binding assays for binding affinity. If strong affinity between gp120 and the glycodendrimers is established, then these glycodendrimers will be further v assessed in an inhibition of viral infectivity assay. This assay will involve HIV in vitro with active viral particles. In the end, this research could lead to the prevention of HIV infection. ________________________________________, Committee Chair Katherine Dawn McReynolds, Ph.D. ______________________________ Date vi ACKNOWLEDGMENTS First I have to thank God, because if it were not for all the signs from above, I would not have completed this difficult task to begin with. There were many times when I needed the inspiration to continue on this journey and my prayers never went unanswered. Next, I must thank my friend and mentor, Dr. Katherine McReynolds, who has challenged, supported, and guided me through this whole process. Without her constant guidance through the Masters program, I believe I surely would have pulled out all of my hair by now. I also want to thank my committee, Dr. Mary McCarthy and Dr. Cynthia Kellen-Yuen, their effort and patience during this tedious writing process has been greatly appreciated. I want to thank all the people in Dr. McReynolds’ research group. Some group members that I particularly want to thank are: Michelle Watterson, Russ Clayton, and Carolyn Lozo, for being my lab buddy when working late nights in the lab. I also want to thank, Vince Trapani, Jon Patane, Careena Cary, and Alex Keith for aiding me in the lab when I needed some extra work done. I also want to thank Janee′ Hardman for testing my sulfated molecule as quick as she could, so that I could mention the results at my thesis defense. I want to say that it has been a pleasure working with each and every one of you. Next, I need to thank the organizations and programs that have aided or led me to vii this point in my education and in research. This includes my tribe, the Duckwater Shoshones. Without their financial support during this time, my advancement in this program would have been incredibly difficult. The Science Transfer Project (STP) and Science Education Equity (SEE) program have provided opportunities that basically led me towards this research. The support the NIH NIAID AREA supplement grant program provided during this time was very much appreciated. I want to acknowledge my parents, Henry and Jeri, whom I love so much. Thank you both for passing on virtues that have helped me in all areas of my life. Finally, I must express my love and gratitude to my husband, Steve Guay. It was my husband that first encouraged me to go back to school and find my niche. Although my major had changed quite a few times along this road, his love, support, and encouragement has always been there. Although hair loss research might have served him better. viii TABLE OF CONTENTS Pages Acknowledgments…………………………………………………………………….... vii List of Figures…………………………………………………………………………… xi List of Schemes…………………………………………………………………….…... xiv Chapter 1. BACKGROUND ………………………………………………………………….......1 Introduction to HIV …………………………………………….…………..…....1 The Structure of HIV-1………………………………………….…………......... 2 Viral Entry and Replication …………………………..…………………..…….. 4 Available Treatments for HIV………..……………….….……..……………..... 8 The Effects of HIV Treatment………………………….…………………….... 13 Developing Research and Treatment for HIV…………………………………. 14 Anionic Sulfated Compounds ……………………..…………..……………..... 18 The Multivalent Effect and Dendrimers ………………………………………. 19 Overview of the Current Project …..…………………………………………... 27 2. RESULTS AND DISCUSSION…………………………………………………….. 32 3. CONCLUSION AND FUTURE WORK ...………………………………………… 84 4. EXPERIMENTAL…………………………………………………………………... 86 Materials............................................................................................................... 86 Instrumentation………………………………………………………………......87 ix Characterization………………………………………………………………….87 Methods..................................................................................................................88 Appendix A. 1H NMR Spectra ........................................................................................102 Appendix B. 13C NMR Spectra .......................................................................................115 Appendix C IR Spectra……………………………………………………………...…128 Appendix D Mass Spectra………………………………………………………..……132 References ……………………………………………………………………………...140 x LIST OF FIGURES Page 1. Illustration of the structure of HIV-1 ...…………………………………………….. 3 2. HIV-1 viral particle seen through electron microscopy…………………………...... 4 3. Mechanism between HIV and the host cell ……………. …………………………. 5 4. Heparan sulfated syndecans and tyrosine sulfated coreceptor CCR5 ……………… 6 5. Overview of HIV replication ………………………………………………………. 8 6. Zidovudine and didanosine …………………………..…………………………….. 9 7. Efavirenz and etravirine ……………….………………………………………….. 10 8. Ritonavir and amprenavir ………….………………………………………...…… 11 9. Enfuvirtide ……………..…………………………………………………………. 12 10. Mechanism of enfuvirtide ………………………………………………..……….. 12 11. Gag assembly that leads to virion maturation ……………………………….……. 16 12. Bevirimat ……………..……….…………………………………………………... 16 13. Heparin sulfate and Dextran sulfate ………………………………………...…….. 19 14. Colominic acid ……………………………………………………………………. 19 15. Divergent and convergent methods ………………………………………………. 21 16. Generations of dendrimers………………………………………………………… 21 17. A generation 2 PAMAM dendrimer …………………………………………….... 22 18. Dendrimer mimic of glutathione peroxidase ………………………………...…….23 19. Generation 4 PAMAM dendrimer………………………………………………… 24 xi 20. (G3- G5) polypropylenimine (DAB-Am) dendrimers………………….…………. 25 21. The dendrimer SPL7013 ………………………………………………….………. 27 22. Hexavalent maltose amino-oxime glycodendrimer ...………….……………….… 29 23. Trivalent maltose amino-amide glycodendrimer ..…...………….…………..……. 30 24. Hexavalent amino core …………….……………………………………………... 31 25. 1 H NMR of Compound 1 …..…………………………………………………...….37 26. 1 H NMR of Compound 3 ……………………………………………………….….40 27. 1 H NMR of Compound 4 ……………………………………………………….… 44 28. An illustration of T1 and T2 ……………………………………………………….. 45 29. IR of Compound 4 ……………………………………...………………...……….46 30. IR of Compound 5….…………...…………………………………………………49 31. 32. 1 H NMR of Compound 6 …………………..…………………………….…..….... 51 Maltose isomers……………………………………………………………………55 33. 1 H NMR of Compound 8 ………………………………………………………..…56 34. 1 H NMR of Compound 9 ……………………………….………………………….58 35. 1 H NMR of Compound 10 ………………..……….……………………………….62 36. Comparing spectra………………………………………………………………… 63 37. MAIDI TOF of Compound 10 ……………………………………..…………….. 66 38. 1 H NMR of Compound 12 …………………..………………………………….….71 39. 1 H NMR of Compound 13 ………………..………………………….…………….73 40. 1 H NMR of Compound 14 …………………..………………………………….….75 41. IR of Compound 15 ……………………………………...………………………..78 xii 42. 43. 44. IR of Compound 16 .………………………………………...………………..….. 80 1 H NMR of Compound 17 .………………………………………………..……… 82 Mass spectrum of Compound 17 ..………………………………………..……… 83 xiii LIST OF SCHEMES Page 1. Synthesis of 3-(3-hydroxypropoxy)propanenitrile, Compound 1 ………..………. 34 2. Example of the Michael reaction …………………………………………………. 34 3. Synthesis of 3-(2-cyanoethoxy)propyl p-toluenebenzene, Compound 3 ………... 38 4. Synthesis of hexavalent nitrile core, Compound 4 ………………...……………... 41 5. Synthesis of hexavalent carboxy core, Compound 6 …………………………….. 47 6. Synthesis of E,Z oxime sugar-linker, Compound 9 ……...…...…….……………..53 7. Synthesis of hexavalent maltose amino-oxime glycodendrimer, Compound 10 .... 60 8. Proposed ring closing mechanism …………………………………………………64 9. Conditions for sulfated glycodendrimer ………………………………………….. 67 10. Synthesis of trivalent amino-amide core, Compound 13 ………….…...…………. 69 11. Synthesis of maltonic acid, Compound 14 ………..……………………………… 74 12. Synthesis of trivalent maltose amino-amide glycodendrimer, Compound 15 ..........76 13. Synthesis of hexavalent amino core, Compound 17 ………..…………...………... 79 xiv 1 Chapter 1 BACKGROUND Introduction to HIV The human immunodeficiency virus (HIV) is a virus that affects millions of people worldwide. In 2009, it was estimated that 33.3 million people were infected with HIV/AIDS (acquired immune deficiency syndrome).1 Within this group there were 2.6 million newly infected cases and 1.8 million deaths as a result of the virus. HIV attacks the immune system and ultimately results in AIDS. The progression from HIV infection to AIDS varies among the infected persons, depending on how each responds to treatments that are available. One available treatment is HAART (highly active antiretroviral therapy).2 In this therapy, a combination of HIV inhibitors are taken together. There are 25 different drugs that are used in various combinations in the HAART regimen. These drugs fall into 5 major categories: protease inhibitors, fusion inhibitors, non-nucleoside reverse transcriptase inhibitors (NNRTIs), and nucleoside reverse transcriptase inhibitors (NRTIs).3 When taken in combination, these drugs have helped many individuals to live with the virus and slow the progression to AIDS. There are some drawbacks though, with the HAART regimen, such as toxic side effects and viral resistance.4 Because of this, more research is needed to overcome these problems. A new area of research to fight against the virus involves attacking it before it enters the 2 host cell.5-7 To achieve this goal, all processes and components of the virus must be evaluated. The Structure of HIV-1 The structural composition of HIV (Figures 1 and 2) has been determined through various studies.8,9 Its appearance is spherical in shape with protruding protein spikes on the outer surface. The protruding components covering the outer membrane consist of a complex of two trimeric proteins, gp120 (glycoprotein, 120 kDa) and gp41 (glycoprotein, 41 kDa). Other components of the viral envelope are lipids and proteins, with the lipids predominating. Within the outer membrane lies the viral matrix, which is composed of viral p17 (protein, 17 kDa). Further inside the virion there is a cone-shaped capsid formed by two thousand copies of protein p24 (a 24 kDa protein). Within the capsid, there are two single strands of viral genomic RNA. There are a total of nine genes in each strand of viral RNA; gag, pol, env, tat, rev, nef, vif, vpr, and vpu. There are also three enzymes in the capsid: reverse transcriptase (RT), protease (PR), and integrase (IN). These collective components of the virion are essential for the invasion and replication of the virus in the host cell.8,9 3 Figure 1. Illustration of one HIV-1 viral particle and its components shown in ribbon form; surface gp120 (SU), transmembrane gp41 (TM), matrix p17 (MA), capsid p24 (CA), nuleocapsid p7 (NC), protease (PR), reverse transcriptase (RT), integrase (IN), and accessory protein (Nef) .8 4 100 nm scale 100 nm scale Figure 2. Ultrathin sectioning was used to visualize this HIV-1 viral particle through electron microscopy.9 Viral Entry and Replication The HIV virus must gain entry into a host cell to replicate. Researchers have discovered that there are multiple entry mechanisms.11 The most common starts with molecular recognition between the virion and the host cell (CD4 lymphocytes, dendritic cells, and macrophages).11 The viral gp120 initially binds to the CD4 receptor on the host cell (Figure 3).12 The binding of these proteins creates a conformational change on the cell surface, which exposes a coreceptor (CCR5 or CXCR4). The co-receptor then binds to a region within the gp120 called the 3rd variable (V3) loop. When this interaction occurs, there is another conformational change within the gp120/gp41 complex and the gp120 dissociates from gp41, allowing the N terminal part of the gp41 (i.e., the “fusion peptide”) to insert into the host cell membrane.10 Following this event, 5 more conformational changes occur and a six-helix bundle is formed within the gp41, causing the viral envelope and the host cell membrane to merge together, resulting in fusion.10-13 (a) (b) (c) Figure 3. The binding mechanism which causes membrane fusion between the HIV virion and the host cell membrane.12 (a) The binding between the viral gp120/41 complex and the host receptor CD4. (b) The interaction between viral gp120 to the CD4 receptor and coreceptor (CCR5 or CXCR4), and the insertion of the N-terminal region of gp41 into the host cell membrane. (c) Gp41 forms a six helix bundle after gp120 dissociates; this process brings the viral and host cell membranes together and fusion occurs .11-14 Another viral entry mechanism involves cell surface glycoproteins. It has been proposed that heparin sulfate proteoglycans (HSPGs) aid in viral entry.15,16 HSPGs are 6 also known as syndecans and are found on all host cell surfaces. Syndecans bind to HIV in the same manner as CCR5 (Figure 4).12 These syndecans are covered with linear polyanionic glycosaminoglycan (GAG) branches which have sulfated sites that interact with the virus. It is thought that the 6-O sulfate groups within the GAG branches are the sites that bind with the viral gp120/V3 loop. Once this binding event occurs there is a second interaction with gp120 within its conserved region (Arg 298).15,16 This process ultimately leads to membrane fusion, allowing viral contents to empty into the host cell so that the replication process can begin.9,14 Figure 4. Syndecans and CCR5 bind to HIV in a similar fashion. The heparan sulfated syndecan (left) has sulfated sugars (top left) that interact with the viral gp120/V3 loop. The CD4 receptor (right) interact with gp120, while the CCR5 coreceptor (right) interacts with the viral gp120/V3 loop with the support of sulfated tyrosine residues (top right) found in CCR5.12,15 7 Viral replication (Figure 5) is a process with numerous steps.11 Once fusion (vide supra) occurs between the target cell and the virus, the viral capsid is released into the cytoplasm of the host cell.9,14 Next, the capsid is uncoated and viral reverse transcriptase starts copying the single stranded viral RNA into double-stranded viral DNA. When this process is complete, the viral DNA is transferred into the nucleus and inserted into the host chromosome by the viral integrase. Following this, transcription begins and viral RNA is created, which then leads to the translation of the RNA to viral polyproteins. The long strands of proteins are cleaved by host and viral proteases, forming viable structural and functional proteins for the new virion. Once a substantial accumulation of these viral components has occurred near the host cell membrane, the budding process begins, forming new virions.9,14 Each time a virion buds, it carries away some of the host cell membrane. Figure 5 demonstrates the replication of one virion.17 In reality, many copies are formed simultaneously. As this phase continues over and over again the host cell membrane is consumed, ultimately killing the host cell. Eventually, the immune system is compromised due to the loss of CD4 positive cells.9 However, scientists have discovered ways to block viral replication before it completely destroys the immune system through drug therapy. 8 Figure 5. An overview of HIV replication. First, viral HIV binds to the host CD4 receptor/coreceptor. Once fusion occurs, the capsid is released into the cytoplasm of the host cell and uncoated. Next, the viral RNA (yellow) is copied into double stranded DNA (red) using the viral enzyme reverse transcriptase. After this, the viral DNA enters the nucleus and is integrated into the host chromosomes (green and yellow) by the viral enzyme integrase. In the nucleus, viral RNA (yellow) is created through transcription. The viral RNA is then transferred out of the nucleus and polypetides are formed during translation. The polypeptides are cleaved by both viral and host proteases into active viral components. The viral proteins and RNA come together and bud from the host cell, forming new virions.9,14,17 Available Treatments for HIV In 2009, the treatment for HIV consisted of 25 drug/drug combinations. These drugs are still being used today and fall into 6 different categories.17 Each class of drugs inhibits a different viral course of action, from viral attachment, to viral protein 9 replication within the host cell. There are five groups of drugs involved in enzymatic inhibition and one group that targets coreceptor/cell entry.17 Starting with enzyme-based inhibitors, there are three groups that target viral reverse transcriptase (RT): nucleoside RT inhibitors (NRTIs), nucleotide RT inhibitors (NtRTIs), and non-nucleoside RT inhibitors (NNRTIs). The NRTIs (zidovudine (AZT), didanosine (ddI), zalcitabine (ddC), stavudine (d4T), lamivudine (3TC), abacavir (ABC), and emtricitabine ((-)FTC)) inhibit the normal nucleotide substrate from binding to the RT active site, and incorporate into viral DNA as chain terminators to stop DNA production. Two nucleoside RT inhibitors, AZT and ddI, can be seen in Figure 6.17 The next RT inhibitor class is the NtRTI group. This group inhibits by incorporating into the 3’ terminus of the viral DNA, terminating the production of viral DNA. There is one widely prescribed NtRTI on the market today, called tenofovir disoproxil fumarate (TDF, Viread®). The last class of RT inhibitors is the NNRTI group. NNRTI (nevirapine, delavirdine, efavirenz, and etravirine, Figure 7) inhibitors bind to an allosteric site on RT, causing enzyme inactivitation.17 Figure 6. Nucleoside RT inhibitors: zidovudine (left), didanosine (right).17 10 Figure 7. Two non-nucleoside RT inhibitors: efavirenz (left), etravirine (right).17 Another class of HIV inhibitors targets viral integrase to prevent the insertion of viral DNA into the host genome.17 Integrase has two important functions, which are 3′processing followed by the strand transfer of viral DNA into the host genome. Before strand transfer takes place, integrase first cleaves viral DNA at the 3′ conserved dinucleotide CA (cytosine adenine). Following this, the complex composed of integrase and the cleaved DNA translocates to the nucleus, where it binds to the host genes in a sequence-dependent manner. There is one integrase inhibitor (INI) that blocks, the strand transfer process called raltegravir.17 The last targeted enzyme within the host cell is viral protease. There are 10 viral protease inhibitors (PIs) available (saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, lopinavir, atazanavir, fosamprenavir, tipranavir and darunavir), all of which compete with the viral polyproteins to bind within the protease active site (Figure 8). Since these PIs 11 cannot be cleaved like the viral polyproteins, they remain in the active site causing inactivity of the enzyme.17 Figure 8. Examples of two protease inhibitors: ritonavir (left), amprenavir (right).17 In addition to the enzymatic inhibitors which work inside the host cell to prevent viral replication, there are two types of inhibitors that work externally to the host cell at the earliest stages of infection. The first class is comprised of fusion inhibitors (FIs). Enfuvirtide (DP-178, pentafuside, T-20, Fuzeon®, Figure 9) is the only fusion inhibitor currently available. It is a polypeptide that blocks the virus from entering the host cell by engaging in a coil-coil interaction with the heptad repeats (HR) of viral gp41, as shown in Figure 10. Unlike the previously discussed drugs, which can be taken orally, enfuvirtide is injected subcutaneously twice a day. This makes enfuvirtide more difficult to use compared to other inhibitors. The second type of inhibitor that works outside the cell are the co-receptor inhibitors (CRIs). Maraviroc, is currently the only one drug that falls into this group. It works on the host cell surface by interacting with the CCR5 or CXCR4 coreceptors, altering their conformation. When this process occurs, there is no molecular 12 recognition between viral gp120 and these co-receptors, so ultimately, viral fusion is prevented.17 Figure 9. The polypeptide enfuvirtide containing 36 amino acids.17 Figure 10. Enfuvirtide (yellow) interacts with viral gp41’s HR1(heptad repeat region 1, red) and HR2 (heptad repeat region 2, blue). This interaction blocks gp41 from folding back on itself, stopping the virion membrane (green) and the host cell membrane (gray) from fusing.17 13 Because the groups of drugs described above are used to inhibit HIV replication via different mechanisms, they are used in combinations as a stronger line of defense against viral invasion.17 Since 1996, drug combinations have been used to fight against HIV. As previously mentioned, this treatment is known as HAART.2,17 This is a regimen that combines at least three anti-HIV drugs from the different classes of inhibitors.17 In the past, some patients were taking more than 20 pills daily. Recently, some HIV inhibitors were combined into one pill, making patient compliance easier. Atripla®, for example, was created in 2006 and contains three different RT inhibitors, tenofovir (NtRTI), efavirenz (NRTI), and emtricitabine (NNRTI).17 Although there are different drugs/drug combinations to combat HIV replication, there are also drawbacks associated with them.2 The Effects of HIV Treatment Since the introduction of HAART, the survival rate for infected patients has risen, but not without some complications. The level of complications encountered depends on how well the patients adhere to treatment, and how far along the disease has progressed before treatment is initiated.2,17-19 When patients follow the recommended regimens, they usually take several drugs a day from at least two different groups of inhibitors. For some patients, the side effects of the drugs are problematic, and they end up not taking the recommended dosage. Other patients are advised to discontinue use of the drugs because of new health conditions arising from the treatment. Various studies have been performed to evaluate the different side effects associated with these antiviral drugs. 14 One study involving 109 patients revealed that the most common side effects were fatigue, stiff and painful joints, aching muscles, diarrhea, depression, and neuropathy.20 The worst problems experienced were changes in physical appearance, through weight gain and lipodystrophy, sleeplessness, joint stiffness and pain, fatigue, and neuropathy. The less severe problems patients experienced were fever and cold-like symptoms.20 Another problem that occurs with the HAART therapy is viral resistance. Viral mutations have been reported with each class of drugs, which leads to inefficient drug therapy for patients.2,21 In the end, when HAART therapy fails, patients have no alternate methods to turn to, therefore new methods are needed to combat HIV. Developing Research and Treatment for HIV Since the discovery of HIV, developing a vaccine has been a very active area of research. Today, the search for a vaccine is still underway; however, some think there is little hope that one will ever be created.22 However, if a vaccine can be created and implemented, some issues involving efficacy, safety, and ethics must first be addressed. The biggest problem is that viral particles may hide in the body undetected in HIV reservoirs, such as the brain, bone marrow, and the lymph nodes.23 Therefore, if a vaccine is administered, its efficiency at preventing infection may not be completely realized.22 The latest vaccine (MRKAd5 HIV-1 gag/pol/nef) created by Merck, was a failure. Their overall results indicated that there was an increase in HIV infection among patients receiving the vaccine compared to the control group in their study.24 While the search for a viable vaccine is still underway, some new anti-viral agents are under development. 15 Inhibition of virion maturation is a new area of research for HIV drug therapy.25 Viral maturation happens after the virus buds from the infected cell, when it matures into a viable particle capable of infection. Scientists are seeking target sites to stop this late stage in viral replication.25 One target is the viral Gag polyprotein, which forms the viral membrane (Figure 11).18 After budding occurs, Gag processing mediated by viral protease continues, which involves the cleavage of the capsid and spacer peptides to form a mature capsid. Recently, it has been found that dimethylsuccinyl betulinic acid (PA457 or bevirimat) may inhibit this processing event in virion maturation.18 While bevirimat (Figure 12) is in phase II clinical trials, some scientists anticipate viral resistance. Because viral resistance has always been a issue with drug therapy researchers are always seeking different avenues to treat HIV. 16 Figure 11. Gag assembly that leads to virion maturation. (A) Composition of Gag: the Nterminal myristate (Myr), matrix (MA) and +++ denotes a highly basic domain, capsid (CA), spacer peptide 1 (SP1), nucleocapsid (NC), spacer peptide 2 (SP2) and the Ctemirmal p6 and Pr55Gag (polyprotein precursor) domains. (B) Illustration of viral assembly, release, and maturation.18 Figure 12. The structure of bevirimat.25 17 Gene therapy is another active area of research that is being investigated to combat HIV. It is known that some individuals are highly resistant to HIV infection, due to a mutation in the CCR5 gene in their CD4 cells. The CCR5 coreceptor works with the CD4 receptor to initiate viral infection (vide supra). Some people do not express the CCR5 coreceptor, due to a 32-base pair deletion in the CCR5 gene.27 If CCR5 is not present on the host cell to aid the CD4 receptor, then the host cell is resistant to HIV infectivity. There is one anecdotal report of apparently successful “gene therapy” based on this. Recently, an HIV patient with acute myeloid leukemia was treated with chemotherapy; the leukemia went into remission but eventually returned. The next line of defense against leukemia for this HIV patient was a bone marrow transplant. The HIV patient was given two bone marrow transplants from a donor who had two copies of this mutated gene. Four years later, his leukemia and HIV have not returned. The successful results of this case has led to more research with the mutated CCR5 gene.26 A small trial was recently performed to mimic the success of this HIV/leukemia patient.26 In the treatment process, the patients undergo leukapheresis to remove their CD4 cells. The CD4 cells are then modified with a zinc-finger nuclease to change the DNA in the CCR5 gene. The mutated CD4 cells are then infused back into the patients. In this study, it was estimated that about 25% of the cells put back into the patients were modified by the enzyme. In some patients these mutated cells persisted for 3 months, but eventually dropped from 25% to just above 5%. As of now, it is not clear if these remaining mutated cells will evolve, but this study has given hope that gene therapy has 18 the potential to cure HIV one day.26 When or if this day will arrive remains unclear. In the meanwhile, researchers are seeking other ways to fight this virus. Anionic Sulfated Compounds It has been established that sulfated polysaccharides have the ability to inhibit HIV.28,29 Two naturally occurring molecules, dextran sulfate and heparin sulfate (Figure 13), have demonstrated the inhibition of HIV-1 infection among CD4 positive cells in vitro, at low (µg/mL) concentrations.28 Inhibition is due to the binding between the sulfated polysaccharides and multiple basic amino acids found in 3rd variable loop region within gp120. Although these compounds displayed inhibitory properties against HIV, there are some significant drawbacks to their use, such as cytotoxicity and increased blood clotting time (anticoagulant effect). One sulfated polysaccharide found to work without these detrimental effects is known as sulfated colominic acid (SCA, Figure 14), an α-28 linked homopolymer of sialic acid. In one study, when compared to dextran sulfate, SCA demonstrated no anti-coagulant activity even at 10 µg/mL, while dextran showed decreased blood clotting at 1 µg/mL.30 It was also found that SCA (6 - 12% sulfur by weight) had more potent anti-viral activity compared to unsulfated colominic acid, and that T-cell growth was not impeded by concentrations up to 100μg/mL.25 Based on these results, some scientists have evaluated SCA further, specifically by combining it with the multivalent effect (vide infra). 19 Figure 13. Structure of two sulfated polysaccharides.5 Figure 14. Sulfated colominic acid.5 The Multivalent Effect and Dendrimers The multivalent effect is a process that improves binding interactions through the attachment of multiple molecules to many receptors at one time.31 Molecules that have this effect are multivalent and have been studied from both natural and synthetic sources. In nature, this process can be illustrated with the influenza virus as it enters the host cell. It has been estimated that between 300 and 600 hemagglutinin spikes on the viral surface 20 allow it to develop a strong interaction with sialyl moieties on the host cell prior to infection.32 This same interaction has been accomplished using a multivalent synthetic compound known as a dendrimer (vide infra).33 Dendrimers are branched macromolecules that are globular in shape and structurally well defined. The main components are the core, the branching structures, and the terminal groups.34 They are assembled either convergently or divergently (Figure 15).35 When a dendrimer is synthesized convergently, the outermost portions are created first then added to a core molecule. The divergent approach consists of building from the core outward. Each method develops a branched system within the compound. Each point of branching leads to a different generation of molecules with an increasing number of reactive ends (Figure 16).35 The multiple termini give it the property of being multivalent. The first class of dendrimers that were created are known as poly(amidoamine), or PAMAM (Figure 17). Since the formation of PAMAM in 1984, the study of dendrimers has become an active research area.34 Dendrimer research is increasing because of the potential these compounds have for molecular recognition and improved binding affinity. Dendrimers could lead to prospective anti-viral compounds, in addition to numerous other applications.34-38 21 Figure 15. Divergent dendrimer synthesis is a method that builds from the core outward. The convergent method combines the branching units and surface groups of the dendrimer together before adding them to the core.35 Figure 16. A generation (G) 7 dendrimer is developed from a poly(amidoamine)-NH3 core.36 22 Figure 17. A generation 2 PAMAM dendrimer.37 Dendrimers are versatile molecules, making them perfect for numerous applications. Because dendrimers can take on different sizes and shapes, they have been synthesized to mimic various molecules.29 Dendrimers have been used as DNA and drug carriers.30,33 They have been created and functionalized to mimic carbohydrate-protein interactions and enzyme-like catalysis as well.29 One example of a dendrimer mimicking enzyme activity was demonstrated by Zhang and co-workers.34 They created a generation 3 dendrimer (Figure 18) that had the catalytic activity resembling glutathione 23 peroxidase, a mammalian enzyme that catalyzes the reduction of hydroperoxides which build up inside the body. Figure 18. Generation 3 dendrimer mimic of glutathione peroxidase.34 Dendrimers also have the potential to inhibit viral infection.33 Since it has been established that the influenza virus binds to a host cell before infection via a multivalent interaction (vide supra), a study was performed to mimic this interaction utilizing a dendrimer.33 The host cell mimic employed in this research was a PAMAM-based dendrimer functionalized with sialic acid residues (Figure 19). This study demonstrated the inhibition of the influenza virus in both in vitro and in vivo. When tested in vitro, the dendrimer blocked viral infection at concentrations of 0.058 - 0.195 mM. When this 24 dendrimer was evaluated in vivo, it displayed no toxic side-effects in the test subjects (14 mice), yet still resulted in the potent inhibition of the influenza virus. The mice were exposed to a lethal dose of the virus mixed with the G4 sialic dendrimer (9 µg per g of body weight). After 14 days there was a 100% survival rate, versus 6% in the control group.33 Through this study, it has been verified that dendrimers have the potential to inhibit viral infection through molecular recognition and the multivalent effect. Because of this, more research has been performed utilizing dendrimers as anti-viral agents.39 OH HO O H N H3C CO2- OH HO H N S H N C G4 PAMAM dendrimer S O Figure 19. Sialic acid conjugated with a generation 4 (G4) PAMAM dendrimer.33 Another study that utilized dendrimer constructs for viral inhibition was performed in 2003.39 This study developed different generation dendrimers functionalized with sulfated galactose residues to inhibit HIV infection. The research demonstrated that the sulfated 3-(β-D-galacto pyranosylthio)-propionic acid derivatives (Figure 20) had the ability to inhibit HIV-1 in CD4 negative cells. Generation 3 - 5 dendrimers had EC50 (effective concentration for 50 % inhibition) values of 90 μM, 70 μM, and 20 μM, respectively. These glycodendrimers also demonstrated no cytotoxicity 25 up to 3 mg/mL. Thus, the dendrimers in this study showed potential to inhibit HIV. After further analysis, the dendrimers described here may become a novel class of antiHIV agents.39 OR OR O H N S RO OR R = H or SO3- O G3-G5 DAB-Am dendrimers Figure 20. 3-(β-D-galactopyranosylthio)-propionic acid with generations 3 thru 5 (G3G5) polypropylenimine (DAB-Am) dendrimers.39 One dendrimer that has been analyzed extensively for its anti-viral properties is SPL7013 (Vivagel®, Figure 21).6,7 This dendrimer has been found to inhibit both HSV (herpes simplex virus)-2 and HIV-1.6 It is now in clinical trials as a microbicide agent. Microbicides are molecules that are used topically, to prevent sexually transmitted diseases. The most recent clinical study performed with the SPL7013 dendrimer involved ex vivo assays. The cervicovaginal fluid (CVF) from 11 women were collected after the vaginal application of one dose of SPL7013 gel.6 The samples were tested with and without seminal fluid over 24 hours for antiviral activity, with both HIV-1 and HSV-2. 26 After a 3 hour period post dose of SPL7013, without the addition of seminal fluid, there was still antiviral activity among samples from all women, with averages of 96% (HIV-1) and 94% (HSV-2) inhibition. After a 24 hour period, 6/11 subjects still demonstrated high antiviral activity with >90% inhibition of both viruses. The assays tested with the seminal fluid were conducted with samples from 3 women. Results revealed similar inhibition values at 3 hours, but the results were mixed at 24 hours, resulting in an overall decrease in inhibition.6 While this dendrimer displayed antiviral activity, more testing is required before it can be used as an HIV inhibitor to prevent HIV infection. Microbicides such as SPL7013 are important in the prevention of the HIV infection. This area of research is just as important as the more traditional treatments for HIV patients. Hopefully, in the near future there will be topical medications available for people to protect themselves from this disease, as well as treatments for people already infected. 27 Figure 21. The dendrimer SPL7013. The divalent benzhydrylamine (BHA) core is in shown in red. The lysine residues are in green, purple, brown, and black, each representing a new branching point, forming a generation 4 dendrimer. The terminal groups in blue are naphthalene disulfonic acid (DNAA).7 Overview of the Current Project There is overwhelming evidence that new HIV inhibitors are needed. Many drugs have been created to combat viral replication, but they are not without drawbacks. Viral resistance and harmful side effects are problems that have led to new research directed towards fighting this virus. Most of the drugs created thus far attack the virus from 28 within an infected host cell. A new area of research to fight against the virus involves attacking it before it enters the host cell. Molecules that have shown the potential to inhibit the virus from entering the host cell are sulfated glycodendrimers. Various studies have demonstrated that there is high binding affinity between sulfated glycodendrimers and viral gp120. Because of these results, more research is necessary in this area. In this study, two glycodendrimers have been created utilizing maltose and tris (2-aminoethyl)amine (tris). Although maltose is not known to have any anti-HIV properties, it is hypothesized that once it is attached to a dendrimer and sulfated, it will exhibit a strong binding affinity to viral gp120 and will inhibit viral infection. Tris(2-aminoethyl)amine was selected to build the cores because of its synthetic versatility and simple structure. Cores of various sizes may be created from the terminal amines, from small and to very large. To create the two glycodendrimers (Figures 22 and 23) utilizing maltose and tris(2-aminoethyl)amine, the following compounds were synthesized: a carboxy-terminated hexavalent core, a trivalent amino core, an oxime sugar-linker, and maltonic acid. An amino hexavalent core (Figure 24) was also created, but was not utilized in this study. The amino hexavalent core will employed at a later date to lend further versatility for future dendrimer construction. Two different methods were employed to create the two glycodendrimers. The convergent process was used to synthesize the hexavalent dendrimer containing six sugars, while the divergent process was employed to create the smaller trivalent dendrimer. Once these glycodendrimers are sulfated, they will be tested in a competitive gp120 binding assay for binding affinity. If significant gp120-glycodendrimer binding 29 affinity is observed, these glycodendrimers will be further evaluated in a viral inhibition assay. In the end, this research could lead to new therapeutic treatments for HIV patients. HO HO HO HO OH O OHO HO O HO HO OH OH HO HO O O O NH HN O O O O HO HO OH OH OH O HO OH HO N N N HO HO N N O OH O O O O O O N N H O O O N H N O HN O O N HOHO HO O OH HO HO O HO OH O O O NH O O N HO HO HO OH O HO O HO O HO OH OH Figure 22. Hexavalent maltose amino-oxime glycodendrimer. N HO HO OH O HO O HO OH OH 30 OH HO OH HO O HO HO OH O OH O HO O OH HO O HO HO HO NH O O O N H O HN HN O N NH O O HN O HO HO HO HO O HO O HO HO HO Figure 23. Trivalent maltose amino-amide glycodendrimer. HO HO 31 NH2 H2N O O N H2N O O N N N O H2N Figure 24. Hexavalent amino core. O H2N NH2 32 Chapter 2 RESULTS AND DISCUSSION The main goal of this research was to create two glycodendrimers that were constructed with the disaccharide maltose and a commercially available amino tris-core. Once created, the glycodendrimers were to be utilized in another study in our lab, by another group member. This included sulfating the glycodendrimers and testing their inhibition properties on a competitive gp120 binding assay. If the sulfated glycodendrimers demonstrate activity, they will then be sent to Duke University, where our collaborator, Dr. Celia LaBranche, would evaluate them for their ability to inhibit HIV infection in human cells. As stated previously, new anti-viral therapies are needed to fight against HIV. Scientists have long known that sulfated molecules have the ability to inhibit HIV infection. The sulfated glycodendrimers created herein have the potential to stop viral infection as well. The compounds created in this study will add to future knowledge directed towards the fight against HIV. Another aim in this study was to employ two different methodologies to synthesize the glycodendrimers. The larger dendrimer (Figure 22) containing six sugars was synthesized using a convergent approach. This method involved building the outer portions of the dendrimer first before attaching them to the core. The smaller dendrimer (Figure 23), containing three sugars was created via the divergent method. In this method, the glycodendrimer was synthesized from the core outwards. 33 The final objective of this research was to add to the library of dendrimer core molecules with an amino hexavalent core (Figure 24). It is important to possess molecules of all shapes and sizes, so a variety glycodendrimers may be eventually created. The amino terminated core is therefore a nice addition. The primary amines on this molecule could be used to add either aldonic acid sugars directly to it, or to create dendrimers of different generations, whereby the primary amines could serve as branching points. All the compounds synthesized to create the glycodendrimers in this study, can be added to our library of molecules and eventually be used in future research to synthesize other glycodendrimers. 3-(3-Hydroxypropoxy)propanenitrile (1) Compounds 1 (the target molecule) and 2 were synthesized via a Michael addition reaction, which is presented in Scheme 1. In a traditional Michael reaction, a nucleophile adds across an olefin or alkyne to produce one major product. An example of this can be seen in a study by Krishna and Jayaraman, in which an alcohol with acryonitrile are combined resulting in a nearly quantitative yield of a single product (Scheme 2).40 The reaction reported here stems from their study, but has been adjusted to fit a diol versus a mono-alcohol. 34 O OH NC HO OH Compound 1 74% Yield + CN 40% NaOH + Acrylonitrile 1, 3 - propanediol NC CN O O Compound 2 7% Yield Scheme 1: Ethereal nitrile linkers (1 and 2) prepared via a Michael reaction, using 1,3-propanediol and acrylonitrile. OH + benzyl alcohol CN acrylonitirle 40% NaOH BnO CN O-benzyl-2-cyano ethanol 99% Yield Scheme 2: Benzyl alcohol and acrylonitrile employed in a Michael reaction to create Obenzyl-2-cyano ethanol.40 In Scheme 1, two nucleophilic sites exist in 1,3-propanediol, creating two different products (Compounds 1 and 2) in the reaction with acrylonitrile. To reduce the formation of product 2, an excess of the 1,3-propanediol (2.23 equivalents) was used. In addition, the NaOH catalyst (40% w/v) and limiting reagent (acryl onitrile) were added dropwise, while the mixture was stirred vigorously. 35 The reaction was monitored by TLC and stopped after 3 days. Usually TLC is used to monitor the disappearance of the limiting reagent and the formation of new products. In Scheme 1, the limiting reagent is acrylonitrile, which has a low boiling point, so it is difficult to visualize by TLC. It was apparent through TLC that two new products were being formed within the first day, but since it was unknown if the acrylonitrile was completely consumed or not, the reaction time was based on a previous reaction, which utilized similar starting materials.41 In that previous reaction, the yields were greatest during the third day, while the yields for the desired product decreased for reactions run for 2 or 4 days. Therefore, the reaction in Scheme 1 was allowed to run for 3 days also. After the reaction was complete, it was quenched with DI water, and then neutralized with 1 M HCl, followed by lyophilization. The residue was then purified by flash chromatography where Compound 2 eluted first, followed by Compound 1. Compound 1 was isolated with a 74% yield. Compound 1 was characterized by both 1H and 13C NMR spectroscopy. The 1H NMR for Compound 1 can be seen in Figure 25. The chemical shifts of each set of protons in Compound 1 are found in the appropriate range, starting with the apparent pentet at δ 1.7. Looking at Compound 1, this signal corresponds to the methylene protons (2), which is the result of two overlapping triplets from CH2 groups (1) and (3). The next chemical shift at δ 2.5 is CH2 group (5) adjacent the nitrile. Further downfield, the hydroxyl proton can be seen at δ 2.9. Since the most electronegative group within Compound 1 is the hydroxyl group, it stands to reason that the group 36 adjacent to it be the most deshielded and will be downfield of the other signals. Therefore the triplet at δ 3.58 is assigned as the methylene group (1). The triplets at δ 3.53 and δ 3.50 almost overlap; they correspond to CH2 groups (3) and (4). Notice these protons are in very similar chemical environments, causing almost identical chemical shifts. In Figure 25, none of the coupling constants have identical coupling partners with the assigned CH2 groups, but the values are close. The reason why the J-value 6.10 Hz for CH2 group (2) is not identical to the coupling constants (both were J = 6.05 Hz) for CH2 groups (1) and (3) is because it is not a true pentet. The pentet is the result of two overlapping triplets that are not well resolved. The reason CH2 groups (4, J = 6.25 Hz) and (5, J = 6.20 Hz), do not have exact J-values, is mostly likely due to the merging triplets of CH2 groups (3) and (4), distorting the splitting patterns. Further confirmation of Compound 1 was conducted via 13C NMR (See Appendix B). 37 2 5 HO 2, J = 6.10Hz O CN 1 3 4 1, J = 6.05Hz 4, J = 6.25 Hz 3, J = 6.05Hz 5, J = 6.20Hz Figure 25. 1H NMR (500 MHz, CDCl3) of Compound 1. A larger version of this spectrum may be viewed in Appendix A. 3-(2-Cyanoethoxy)propyl p-toluenesulfonate (3) Compound 3 was created under biphasic conditions, as seen in Scheme 3. This reaction was performed because the alcoholic OH on Compound 1 needed to be converted into a better leaving group, for the subsequent reaction. Since tosylates are good leaving groups, tosyl chloride was used in this procedure to synthesize Compound 3, a tosyl ester. 38 CN HO O TsCl, CH2Cl2 50% NaOH, 50°C Compound 1 CN TsO O Compound 3 30.4% Yield Scheme 3. Compound 1 was tosylated under basic conditions to produce Compound 3. In this reaction, vigorous stirring and heating were needed to mix the biphasic solution. First, Compound 1 was dissolved in dichloromethane and heated to 50 degrees Celsius. Next, 2 equivalents of tosyl chloride were added, followed by aqueous NaOH, which was added dropwise. The reaction was stirred and allowed to reflux for 6 days before stopping. The reaction was monitored by TLC, and after the third day there were still visible signs of starting material, Compound 1. Because of this, more NaOH, dichloromethane, and tosyl chloride were added. The temperature was also increased to 54ºC. After Compound 1 was no longer seen by TLC, the reaction was stopped on the sixth day. Once the reaction was removed from heat, the organic phase was extracted and concentrated by rotary evaporation. Next, the residue was purified via flash chromatography. The desired product 3, eluted as the third band, followed by a mixture of Compounds 1 and 3. The yield for pure Compound 3 was 30%. Compound 3 was characterized by 1H and 13C NMR spectroscopy. 39 The 1H NMR for Compound 3 can be seen in Figure 26. The doublets at δ 7.7 and δ 7.3, which arise from the tosyl group, display a para-substitution pattern for an aromatic ring. The doublet at δ 7.7 corresponds to the CH groups at position (4, 5) since they are near the tosyl SO3 electron withdrawing group. The doublet at δ 7.3 relates to the two protons at position (2, 3), which are adjacent a methyl group, moving them slightly upfield. As seen in Figure 26, groups that couple with identical J-values are CH2 groups (7) and (8) with J-value 5.95 Hz and CH2 groups (9) and (10) with the coupling constant 6.35 Hz. The last set of CH2 groups (6) and (7) couple with J-values 6.00 Hz and 5.95 Hz, respectively. The remaining singlet at 2.4 ppm, corresponds to the methyl group at position (1), which has no neighboring protons, therefore appears as a singlet. Compound 3 was also characterized via a 13C NMR (Appendix B). 40 2 4,5 J = 8.25Hz 5 O 1 2,3 J = 7.95Hz S O 3 4 7 O 10 O CN 6 8 9 1 9, J = 6.35Hz 8, J = 5.95Hz 10, J = 6.35Hz 7, J = 5.95Hz 6, J = 6.00Hz Figure 26. 1H NMR (500 MHz, CDCl3) of Compound 3. This spectrum may also be seen expanded in Appendix A. It should be noted that the reaction in Scheme 3 was performed numerous times and yields were low in each trial. At first it was thought that yields were low due to the biphasic system. Because of this pyridine was employed, replacing the aqueous NaOH, in order to create a more homogenous mixture. Through 1H NMR it was apparent that product formation was actually worse using pyridine, so the residue was not completely purified and no yields were calculated. To increase yields in the future, a few variables could be changed to create Compound 3. In a previous reaction similar to the procedure in Scheme 3, the heat was increased to 90ºC by mistake for a short period and the yield reached 51.5%. This is one 41 variable that could be investigated to increase yields. Due to time constraints, this was not performed in this study. Hexavalent nitrile core (4) The hexavalent nitrile core 4, which can be seen in Scheme 4, was created for multiple reasons. The terminal nitrile groups on the hexavalent core 4 make it very versatile, since it could be hydrolyzed to create a carboxy core or reduced to form an amine-terminated core. CN O CN NC O O H2N TsO O CN + N NH2 N K2CO3, CH3CN N N O 90°C Compound 3 CN N NH2 Tris(2-aminoethyl)amine O NC O Compound 4 91% Yield CN Scheme 4: Six equivalents of tosyl linker 3 were added to the commercially available tris(2-aminoethyl)amine to form the hexavalent core 4. To perform the reaction seen in Scheme 4, a very dry environment was required. All glassware used was flame-dried and placed under nitrogen gas before adding any starting materials or reagents. Once the acetonitrile and potassium carbonate were added together, the reaction was stirred and heated to 89ºC. Following this, tris(2-aminoethly) 42 amine was added to the mix. Then a combination of the tosyl nitrile linker 3 (7.67 equivalents) in acetonitrile was added in portions, over a four hour period. Over the next 24 hours, the heat was lowered to 82ºC and allowed to stir at this temperature until stopped. The reaction was halted after two days, since a similar reaction done by DeCampo and co-workers was found to be optimize in that time frame.42 TLC was performed to analyze reaction progress. Due to the polarity of the limiting reagent (tris 2-(aminoethyl) amine) and the desired product (Compound 4), it was difficult to verify the completeness of the reaction. It was apparent that there was product formation after the first day, and after the second day it appeared that the tris(2-aminoethyl)amine was consumed. Because both the tris-core and hexavalent core 4 have similar Rf values with any solvent system, the reaction was stopped based on a color change in the reaction spot on the TLC plate. When the color for the tris-core, which was yellowish, had disappeared from view on the TLC plate, the reaction was thought to be complete. This happened after the second day, which coincides with the reaction done by DeCampo and co-workers, which was also complete in two days.42 Once the reaction was stopped, it was filtered and condensed in vacuo. Next, an extraction was performed on the residue and the resulting solution was concentrated through evaporation. Since the hexavalent core 4 was found to be amphiphilic, the excess tosyl-linker 3 was removed using a water rinse. The tosic acid formed in this reaction was removed via a batch rinse with cationic resin. A 91.1% yield was obtained 43 for Compound 4, which was characterized by, 1H and 13C NMR, as well as mass spectrometry. The 1H NMR for Compound 4 is given in Figure 27. Looking at this spectrum, the three peaks furthest downfield at δ 3.59, δ 3.47, and δ 2.56 are sharp and well-defined triplets, whereas the chemical shifts at δ 2.46 and δ 1.66 are less clear. The reason for both the poor integration and broad and an unclear splitting patterns has to do with the size of the molecule and the relaxation times in the NMR experiment.43 Because Compound 4 is a large molecule, it tumbles slowly in solution. The inner atoms have limited motion and are not able to interact with neighboring nuclei very well. This causes an increase in the time it takes for the nuclei to relax back to their original orientation after having been pulsed with energy during an NMR experiment.44 Nuclei in a magnetic field precess randomly about the magnet in different spin states until a radio frequency pulse is applied. When energy is absorbed, nuclei change from a low energy spin state to a high energy spin state and the nuclei become more organized as they precess in their spin states about the magnetic field (Figure 28).45 When the nuclei release this energy, they return (relax) back to their original orientation. The time it takes for the nucleus to relax (i.e. emit energy) can be broken up into two components, longitudinal relaxation time (T1) and transverse relaxation time (T2). T1 corresponds to the time it takes for nuclei to return to their original spin state as they release energy to neighboring nuclei. T2 corresponds to the amount of time it takes for organized precessing nuclei to return to their original axis of rotation about the magnetic field.43,44 T1 and T2 relaxation times affect the NMR signal as energy is released. T1 affects the intensity of the observed 44 NMR signal and thus the integration of the peaks, while T2 affects the width of the peaks. In large molecules both relaxation times may be affected due to the increased time it takes to relax, resulting in line broadening.43 CN 6 5 3 7 O 1 4 N N 2 O 3 7 5 6 CN 3 5,6 J = 6.27Hz 7, J = 6.21Hz J = 6.27Hz 1,2,3 4 Figure 27. 1H NMR (300 MHz, CDCl3) of Compound 4. An extended spectrum of this NMR may be viewed in Appendix A. 45 T1 T2 Figure 28. An illustration of T1 and T2 in a NMR experiment. T1 (left) represents the different energy spin states. T2 (right) demonstrates nuclei precessing about the magnetic field (a) before being pulsed with energy and (b) after irradiation.45 In Figure 27, it is apparent which chemical shifts have been affected by the relaxation time. Looking at Compound 4, the CH2 groups that are more susceptible to line broadening are (1-4), which are buried within this molecule. The broad apparent pentet at δ 1.66 is assigned to CH2 group (4) since it is the most shielded CH2 group due to the adjacent methylene groups. This leaves CH2 groups (1-3) which gives the broad signal at δ 2.46. The triplet at δ 2.56 was assigned to CH2 group (7) because it is adjacent a nitrile. The last two triplets between δ 3.47 and δ 3.59 were assigned to CH2 groups (5-6) since they are adjacent the electronegative oxygen atom. Since CH2 groups (5) and (6) have identical coupling constants, it is unclear which couples to CH2 group (7). Further confirmation of Compound 4 can be viewed in the 13C NMR (Appendix B) and the IR (Figure 29, Appendix C) spectra. The IR shows a nitrile peak at 2251 cm-1 (Figure 29). This aspect will be important later when discussing the hydrolysis of Compound 4. A matrix assisted laser desorption ionization-time of flight mass 46 spectrometry (MALDI-TOF MS) spectrum was also collected for Compound 4 (Appendix D). 87.2 80 70 1667.75 60 1328.21 1414.44 50 844.40 951.82 Nitrile 665.83 %T 40 3017.84 2251.04 30 CN 1467.07 1224.85 O 20 N 1367.96 N O 10 CN 2873.23 3 755.07 1118.35 -2.0 4000.0 3000 2000 1500 1000 600.0 cm-1 Figure 29. IR of Compound 4, with a nitrile peak at 2251 cm-1. This spectrum can be seen in Appendix C in an expanded view. Methyl ester core (5) As can be seen in Scheme 5, Compound 5 is an intermediate, which was synthesized in order to reduce the overall salt formation that results from the direct hydrolysis of Compound 4 to a carboxylic acid. In prior reactions that were basecatalyzed, the salt formation was extensive and it was time consuming to remove, as the 47 target Compound 6 was also completely water soluble. Therefore, to minimize salt formation, an acid-catalyzed reaction was employed as the first step in Scheme 5, in which hydrochloric acid is created in situ utilizing acetyl chloride and methanol. O CN O O CN O O O O O Acetyl chloride, MeOH N N O CN 0°C O 25°C N O N O N N O N O CN O O N O NC O O Compound 4 O Compound 5 O 33% Yield NC O O O LiOH, H2O, mw O OH O OH O O HO O N O N N HO O N O O O Compound 6 67% Yield O OH O OH Scheme 5: The hexavalent core 4 undergoes esterification in the first step to form the hexa-methoxy Compound 5. Hydrolysis takes place in the second step to form carboxylic acid Compound 6. 48 This esterification reaction commenced with the combination of the hexavalent nitrile core 4 and dry methanol. The mixture was cooled in an ice bath before adding the acetyl chloride to keep the hydrochloric acid in the solution upon formation.46 The mixture was stirred overnight at 25ºC. Due to the similar polarities of both Compound 4 and Compound 5, the reaction was analyzed with colorimetry via TLC. Since these compounds appeared different when stained with ninhydrin, it was used to differentiate between both products. Starting material 4 had appeared brown by TLC using ninhydrin stain, while Compound 5 looked yellowish. When Compound 4 was no longer visible, the reaction was stopped. After 24 hours was concentrated in vacuo and then freeze dried overnight. Next, to remove any unreacted starting material or partially formed products, dialysis was performed. It had been established previously that the starting material, Compound 4, is not retained while in dialysis tubing. During one trial, when Compound 4 was purified by dialysis, using 100 MWCO (molecular weight cut-off) tubing, some product was found to have gone through the tubing.41 It should be noted that the methyl ester 5 may also have the ability to escape the dialysis tubing. This may explain the low yields. After the sample remaining in the 100 MWCO tubing was freeze dried, it was analyzed by NMR spectroscopy. The 1H and 13C NMRs performed on Compound 5 were inconclusive. Multiple samples were analyzed by NMR spectroscopy, using both CD3OD and D2O solvents. During some NMR experiments, the NMR would not even lock and at other times when a signal was obtained, there was extreme peak broadening (vide supra) and integrations 49 could not be attained. Because of these difficulties Compound 5 was analyzed by IR (Figure 30) instead. In this spectrum, a strong ester carbonyl stretch was observed at 1733 cm-1, and the sharp nitrile peak at 2254 cm-1 (Figure 29) was no longer visible, indicating the successful production of Compound 5. This product was carried forward without any further purification. 83.6 80 75 70 2924.15 %T 65 O O O 60 N 1732.95 Ester carbonyl 1197.27 1112.88 N O 55 O O 3 50.0 4000.0 3000 2000 1500 1000 cm-1 Figure 30. IR of the Compound 5 displays a strong ester carbonyl peak at 1733 cm-1. An expanded version of this spectrum may be viewed in Appendix C. Hexavalent carboxy core (6) The hexavalent core was next functionalized with terminal carboxylic acids to assist in amide coupling which are used to create the large glycodendrimers in this research. Compound 6 was created from the second reaction shown in Scheme 5. This 400.0 50 ester hydrolysis reaction was performed in a microwave to reduce the reaction times from days to minutes.41,47 To synthesize Compound 6, the methyl ester 5 and nanopure water were combined, followed by the addition of lithium hydroxide. The mixture was placed in a microwave and heated for 25 minutes at a power setting of 400W. The first 2 minutes in this process were used to ramp the temperature slowly up to 60ºC, where the reaction was held for the remainder of the reaction time. After the mixture was heated for 23 more minutes, it was cooled to room temperature. When the reaction had cooled to room temperature, it was concentrated in vacuo to remove the water. The sample was then reconstituted in methanol, stirred, then centrifuged to remove the leftover lithium hydroxide. This was followed by size exclusion FPLC (fast paced liquid chromatography), which was performed to remove any side products and any remaining salt. A strong signal at 214 nm was observed on the chromatogram, indicating the formation of the desired product, Compound 6. The peaks that demonstrated an absorbance at 214 nm were collected and resulted in 66% yield (68.1 mg) of Compound 6. The identification of Compound 6 was verified through first by NMR spectroscopy and then mass spectrometry. The 1H NMR spectrum for Compound 6 is given in Figure 31. The spectrum demonstrates peak broadening, due to the slow relaxation times (vide supra) for Compound 6. The peaks at δ 1.80-1.90 and δ 2.75-2.90 are very broad and indistinct splitting patterns. The signal at δ 3.58 displays some peak broadening as well. The two well-defined peaks at δ 2.45 and δ 3.71 were assigned to CH2 groups (7) and (6), 51 respectively, since they have the same coupling constant (J = 6.60 Hz) and are positioned in the appropriate area. The other CH2 groups were assigned by their expected chemical shifts. Methylene (4) would be the most shielded and upfield, and while CH2 groups (13) are expected to have similar polarities and therefore should appear at similar chemical shifts. Assignments were further confirmed by integration of the assigned peaks, which added up to the correct proton count needed for Compound 6, as can be seen in Figure 31. Characteristics of Compound 6 can also be seen in the 13C NMR in Appendix B. Compound 6 was additionally analyzed by time of flight negative mode electrospray mass spectrometry (TOF ES- MS) available for view in Appendix D. O 6 5 3 OH O 7 1 4 N N 2 5 6, J = 6.60Hz 7 O 3 OH 6 O 3 7, J = 6.60Hz 5, J = 6.10Hz 1,2,3 4 Figure 31. 1H NMR (500 MHz, D2O) of Compound 6. An extended view of this spectrum may be found in Appendix A. 52 Boc-protected oxime sugar-linker (8) Compound 8 was synthesized under acidic conditions (Scheme 6). Since this was a condensation reaction in which water was formed through the coupling of maltose and Compound 7 (made previously by a group member), keeping free acidic protons available in the reaction was important. The buffer used here was 0.1M ammonium acetate (pH 4.5). The Boc-protected linker 7 used in this reaction was employed for two reasons. The first was for purification purposes. Creating a less polar molecule than maltose would make separation easier via flash chromatography. The second reason was to make characterization obvious. The Boc-protecting group creates a unique singlet around δ 1.40 that integrates for 9H in 1H NMR spectroscopy. 53 OH O HO HO OH HO + O O HO Maltose H2N H N O O Boc Compound 7 HO OH Buffer NH4OAc (4.5) OH O HO HO OH HO O OH O N HO O Boc HO Compound 8 76% Yield TFA CH2Cl2 OH HO HO H N O OH HO O OH HO O N NH3+ O HO Compound 9 Quantitative Yield Scheme 6: The coupling of maltose and a Boc-protected amino Compound 7 under acidic conditions to form Compound 8, followed by a deprotection step creating an amino oxime sugar-linker 9. Maltose, Compound 7, and an ammonium acetate buffer were combined, and the pH was monitored via pH paper. The pH remained at ~4.5 during the entire reaction. The reaction was also monitored by TLC. When Compound 7 disappeared from view, the reaction was stopped and the sample was lyophilized. The sample was purified further through flash chromatography. Unreacted Compound 7 eluted first, then the 54 desired product (Compound 8), followed by maltose. Compound 8 was collected and concentrated by rotary evaporation. As expected, the Boc-protecting group did promote separation from maltose, yielding 76% of Compound 8. Identification of Compound 8 with the Boc was also apparent as seen through 1H and 13C NMR spectroscopy. Before the analysis of Compound 8 by 1H NMR spectroscopy can be examined, the NMR properties of maltose should be discussed. Because maltose (Figure 32) comes in two forms, there are distinctive chemical shifts for each structure. In α-maltose, there are two anomeric protons in the equatorial positions that form two doublets at δ 5.24 and δ5.42. In β-maltose, there is one anomeric proton in the axial and one in the equatorial position at δ 4.67 and δ 5.42, respectively. When maltose is coupled to another molecule, the doublet corresponding to the non-reducing anomeric proton should always be visible downfield, left of the D2O solvent peak. The other sugar hydrogens overlap, creating a complex splitting pattern which will always be apparent between 3.20 and 4.20 ppm. 55 OH OH O O HO HO HO HO H' OH HO O O OH HO H Maltose OH HO O O HO H' HO Maltose OH HO H D2O H' H J = 3.81Hz H J = 7.95Hz J = 3.78Hz Figure 32. 1H NMR (300 MHz, D2O) of Maltose. α-Maltose with two equatorial positioned protons in red (H' = unreactive proton) and β-maltose with one axial proton (green) and one equatorial proton (red) emphasized. Looking at the 1H NMR spectrum for Compound 8 (Figure 33), the doublets that appear furthest downfield at δ 7.60 and δ 7.00 correspond to the vinylic hydrogens of the E and Z stereoisomers.48 The chemical shift at δ 7.60 arises from the E isomer, since this isomer (trans) is more stable then the Z (cis) isomer, and therefore should be seen in larger quantity. As can been seen in Figure 33, the signal corresponding to the E proton (δ 7.60) is stronger than the Z proton peak (δ 7.00), demonstrating the production of more of the E isomer Compound 8, in a 6:1 ratio. The next major signal left of the HOD peak in Figure 33 relates to the nonreducing anomeric proton. As stated previously there 56 should always be a doublet in this area (above 4.80 ppm) representing this proton. The other peaks ranging from δ 5.40 to δ 3.40 correspond to sugar hydrogens and some CH2 groups (3-5). Since these groups overlap, coupling constants could not be measured. The peaks that did exhibit clear coupling were seen at δ 3.14 and δ 1.78 which correspond to CH2 groups (7) and (6), respectively. Since CH2 group (6) is more shielded versus the other CH2 groups it was assigned to the most upfield of these two peaks. The peak at δ 1.45 is the signal from the methyl groups on the Boc protecting group. Since these CH3 groups have no neighbors to create a splitting pattern, one singlet is formed. Compound 8 was also analyzed by 13C NMR (see Appendix B). E J = 6.05 Hz 1 Z J = 5.50 Hz OH O HO HO H' HO OH O 7 7 OH 2 O N O HO HO 3 H N 5 4 O 7 6 O HE,Z 7 D2O 1,2-4 1 J = 3.90 Hz 6, J = 6.40Hz 5, J = 6.45Hz H' Figure 33. 1H NMR (500 MHz, D2O) of Compound 8. This spectrum may also be viewed (expanded) in Appendix A. 57 E,Z oxime sugar-linker (9) The Boc-protecting group was removed from Compound 8 by the addition of TFA (Scheme 6). The reaction was analyzed by TLC using ninhydrin stain. It was evident that the reaction was complete after 3 hours by TLC, as indicated by a color change from the light pink spot of the starting material being replaced by a strong purplepink band. When the reaction was complete, all solvents were removed through evaporation and lyophilization. After the sample was freeze dried, it was dissolved in ammonium bicarbonate (0.03M) instead of water. This was to help remove TFA that was not removed during rotary evaporation. The yield of Compound 9 was quantitative. The product was identified by 1H NMR (Figure 34), 13C NMR (see Appendix B), and Mass spectrometry (see Appendix D). The 1H NMR spectrum for Compound 9 is presented in Figure 34. Notice that the singlet corresponding to the removal of the Boc group at 1.45 ppm is absent. This is the only real difference between Figures 33 and 34, except that the coupling constants for CH2 groups (6) and (7) could no longer be measured. 58 1 J = 5.95 Hz E J = 5.55 Hz Z OH O H' HO HO OH OH HO O HO 2 5 O N O 3 HO 4 NH3+ 6 HE,Z D2O 1,2-4 1 J = 3.85 Hz 6 5 J = 3.90 Hz H' J = 5.15 Hz Figure 34. 1H NMR (500 MHz, D2O) of Compound 9. This spectrum may also be viewed (expanded) in Appendix A. Hexavalent maltose amino-oxime glycodendrimer (10) Compound 6 and the oxime sugar-linker Compound 9 were coupled through amide bond formation under dry conditions (Scheme 7). Because Compounds 9 and 10 are both hydrophilic, they were dissolved together in nanopure water and lyophilized overnight. When they were removed from the freeze dryer, they were immediately placed under nitrogen gas. Six sugar-linkers 9 were needed per one carboxy core 10, thus 6.3 equivalents of the Compound 9 were used. After the solvent dimethyl sulfoxide (DMSO) was added, O-(benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) was mixed into the reaction. TBTU was added in excess (14.4 equivalents) in 59 order to neutralize any residual TFA left from the formation of the sugar linker 9. It was recommended to add at least 1.2 equivalents of TBTU for every carboxylic acid,49 so 7.2 equivalents were added for Compound 10 and 7.2 for any remaining TFA. The base, N,N-diisopropylethylamine (DIPEA) was added last to a pH of 8.5. Basic conditions were important in this reaction to activate TBTU. DIPEA was added in portions of 100 μL and allowed to stir for ~ 30 minutes before checking the pH, using wet pH paper. After 300 μL were added, the desired pH (8-9) was reached. The reaction was allowed to stir for 24 hours before stopping. This reaction was stopped based on the work done by Baek and Roy.49 In their work, product formation was complete within 24 hours as shown by a negative ninhydrin test upon TLC. This negative ninhydrin test would not work for this reaction, because the limiting reactant here is not the amine Compound 9, which would always give a positive result. Even so, the reaction in this study was stopped at 24 hours, since Baek and Roy’s reaction was complete in this amount of time.49 60 O HO OH O OH O O HO HO O N HO O N O HO O HO OH + O N OH OH TFA- N NH3+ O O HO Compound 9 O N Compound 6 O O O TBTU, DIPEA, DMSO, N2 O OH OH HO HO OH O HO O HO OH OH N O O H N O HO O Compound 10 66% Yield OH O HO HO HO OH OH O HO HO N N O N O O N H O 3 Scheme 7: Glycodendrimer 10 formed through the coupling of 6 and 9. When the reaction was stopped, it was first concentrated by rotary evaporation. Next, a large volume of water was added to the reaction mixture and it was lyophilized overnight to remove any remaining DMSO. The following day, an extraction was performed to remove any side-products that were organic soluble. The aqueous phase was freeze dried again. After lyophilization, a white precipitate (ppt.) formed in the sample when it was dissolved back into water, so filtration was performed on the sample. The ppt. collected was not analyzed and discarded because it was thought to be TBTU byproducts. This conclusion was based on previous trials using a similar reagent BOP 61 (benzotriazol-1-yloxy-tris(dimethylamino) phosphonium hexafluorophosphate), which reacted the same way after being freeze dried. Compound 10 was then further purified by dialysis. Since the desired product 10 had a molecular weight (MW) of 3567 g/mol, the dialysis membrane used had a 2000 molecular weight cut off (MWCO). After 7.5 hours of dialysis, a fluffy yellow-orange solid was recovered through lyophilization and analyzed by 1H and 13CNMR spectroscopy. Given that Compound 10 is a large molecule (exact mass = 3567.73 g/mol) most peaks in its 1H NMR (Figure 35) were broad due to long relaxation times (vide supra). To get a more accurate proton count, only one arm of the dendrimer was integrated. The observed (38) and expected (38) proton count were a match after this spectrum was integrated. Even though the peaks were not well-defined in Figure 35, all the CHn groups were assigned by comparing starting materials to the product. A comparison spectrum can be seen in Figure 36. The two peaks between δ 1.80-2.10 correspond to CH2 groups (6) and (11), the most shielded protons. The signal at δ 2.50 relates to CH2 group (8), which is adjacent to the carbonyl group. Next, the very broad peak between δ 2.80-3.20 was assigned to CH2 groups (12-14), which were the protons most hidden within the core. The chemical shift at δ 3.25 was assigned to CH2 group (7), and the peaks spanning from δ 3.30 to 5.40 were assigned to CH2 groups from the sugar-linker (1,3, and 5) and groups (9) and (10) from the core. The last two peaks at δ 7.60 and δ 7.00 corresponded to the E and Z oxime protons. Again, the E proton was assigned to the more intense doublet (vide supra). The intensity for both signals have decreased though, which was most likely due to the closing of the sugar ring.48 62 J = 5.95 Hz E J = 5.40 Hz Z 1 HO HO OH O H' HO OH OH O HO HO H N 5 2 N O O 4 3 7 8 6 O HE,Z 10 O 9 13 N 11 N 12 3 2 1-4,8,9 1 J = 3.70 Hz 6 11-13 7 5,10 H' Figure 35. 1H NMR (500 MHz, D2O) of Compound 10. This spectrum may also be viewed (expanded) in Appendix A. 63 Figure 36. A spectra comparison of: (top), the hexavalent carboxy core 6, (middle), the hexavalent amino-oxime glycodendrimer 10, (bottom) of the E,Z oxime sugar-linker 9. Ring closure is a phenomenon that occurs among carbohydrates. Scheme 8 demonstrates a proposed mechanism of the oxime formation and the ring closing of Compound 8.48 In this scheme, after maltose is placed under mildly acidic conditions (pH 4 - 5), it is oxidized into an open chain aldehyde (aldehydo). Next, the electrophilic aldehydo is attacked by the primary amine end of Compound 7 forming the carbinal oxyamine intermediate. When the intermediate loses water, Compound 8 is formed. The last step in Scheme 8 demonstrates the two structures of Compound 8 that seem to appear to be in equilibrium by 1H NMR spectroscopy. It has been found that over time, 64 all of the oxime Compounds 8, 9, and 10 start to undergo ring closure as they are being analyzed by 1H NMR spectroscopy. This explains the decreased signal intensity for the E and Z isomer compounds. OH O OH HO HO OH HO O HO Maltose Buffer NH4OAc (4.5) aldehydo O HO HO O OH HO HO OH O HO OH O HO H H2N H N O O Boc Compound 7 OH O HO HO OH HO O OH OH HO HO Oxyamine intermediate H H N O Boc O N H - H2O OH O HO HO OH HO O OH N HO O H N O HO Boc H Compound 8 H2O OH O HO HO OH HO O O H N HO O O HO H Scheme 8: Proposed oxime formation and ring closing mechanism.48 H N Boc 65 The 13C NMR of Compound 10 can be viewed in Appendix B. The next analysis performed on Compound 10 was matrix assisted laser desorption-ionization time of flight mass spectrometry (MALDI-TOF MS, Figure 37). The peaks seen at 3622.790 m/z and 3640.374 m/z are the mass of Compound 10 with the addition of water. The mass 3622.790 is [M+H+3H2O] and the mass 3640.374 is [M+H+4H2O]. Compound 10 [M+H] = 3568.735 is a close match to the peak at 3570.280 m/z. Because this is a large molecule, the mass may vary due to a mixture of isotopes. Since Compound 10 has 144 carbons, there is most likely some 13C present, causing an increase in the mass. After Compound 10 was characterized in this study, it carried forward in another study in our lab. 66 [M+H] C144H270N16O84 Exact Mass: 3567.73 1 HO HO OH O HO O HO OH OH N O H N O N N O HO O 2 3 [M+H+4H2O] [M+H+3H2O] Figure 37. MALDI-TOF of Compound 10. This spectrum may also be viewed expanded in Appendix D. The ultimate purpose for the synthesis of Compound 10 was to test it for any anti-viral properties. Therefore, once Compound 10 was synthesized, it was sulfonated (Scheme 9) to maximize its inhibition potential.50 This sulfated Compound 10 has been sent off for an elemental analysis. It has been determined by Columbia Analytical Services in Tucson Arizona, that Compound 10 has a sulfur percentage of 12.93%. This measures out between 21 or 22 sulfate groups on Compound 10, translating to nearly 2 sulfate groups per sugar. Soon it will be evaluated for its binding affinity to the HIV 67 protein gp120 in a competitive binding assay. The other glycodendrimer created in this study will eventually be tested in the same fashion. HO HO OH O HO O HO OH OH N O O H N O HO O Compound 10 N OH O HO HO HO OH OH O HO N O N O O N H HO O 3 SO3-pyridine DMF, N2, 0°C 1 hour RO RO OR O RO O RO OR OR N O O H N O RO O Sulfated Compound 10 OR O RO RO RO OR OR O RO N N O N O RO O N H R = H or SO3- Scheme 9. Conditions to sulfate glycodendrimer 10.50 O 3 68 Synthesis of the Boc-protected trivalent amino amide core (12) To create the glycodendrimer containing three disaccharides, a trivalent core was needed. To begin the process, Compound 12 was created from the Boc-protected linker 11 (which was made previously by a group member) and tris(2-aminoethyl)amine (Scheme 10). Compound 11 was freeze-dried beforehand to remove moisture. After 11 and tris(2-aminoethyl)amine were combined, they were immediately placed under nitrogen gas. The following reactants were then added sequentially: BOP, DMF, and DIPEA. BOP promotes the amide coupling of 11 and tris(2-aminoethyl)amine, therefore an excess was used. After the components were dissolved in DMF, DIPEA was added and the reaction was allowed to mix for three days at which time the reaction was stopped based on the results of a previous trial. 69 NH2 N O H N O N H2N NH2 N H BOP, DIPEA,DMF, O Tris (2-aminoethyl) amine N2, 0°C O Compound 12 27% Yield RT 3 + O HO2C TFA CH2Cl2 Boc N H Compound 11 TFAH N O NH3+ N O Compound 13 Quantitative Yield 3 Scheme 10: The formation of Boc-protected core 12 using the commercially available tris-core and a previously synthesized carboxylic acid 11. In the second step, the trivalent amino core 13 was created by the removal of the Boc protecting group. When the coupling reaction was stopped, the reaction was concentrated in vacuo, followed by three extractions. They were performed in succession to help remove BOP byproducts and DIPEA. The sample was concentrated by evaporation and lyophilized overnight. The organic phase was dried with anhydrous sodium sulfate overnight, followed by purification via flash chromatography. Three flash columns were necessitated for this sample due to the BOP byproducts and leftover Compound 11. In the first separation, the solvent system was 8:1 chloroform:methanol. This step removed leftover linker 11. The second flash column was performed to elute BOP byproduct first with less polar solvents. After the residue was added to the column it was eluted first with ethyl acetate, then ethyl acetate:methanol (95:5). Lastly, when methanol alone was 70 added the product eluted. Although this process did remove some BOP byproducts, some impurities remained, so, for the last flash column, a shorter silica plug was employed. The same solvents were used as above and a pure sample of Compound 12 (27% yield) was collected after the BOP byproduct eluted. This was confirmed by NMR spectroscopy. When the 1H NMR (Figure 38) for Compound 12 was integrated, there was almost a perfect match of 63 protons even though some peak broadening (vide supra) was visible. The peaks relating to the amide hydrogens are seen at δ 5.30 and 7.10. The broad peak at δ 3.30 was assigned to CH2 groups (1) and (2), since they are protons within the core structure. The broad peak at δ 2.58 and the triplet at δ 2.43 were assigned to CH2 groups (4) and (7). CH2 group (4) was assigned to this area (δ 2.43 and δ 2.58) because protons adjacent a carbonyl group were found in this range. CH2 group (7) was also assigned in this region, because once Compound 12 was deprotected, a peak in this area disappeared, which most likely corresponded to CH2 group (7). The remaining peaks were sharp and well-defined. The sharp singlet at δ 1.40 is indicative of the Boc group. The triplets between δ 3.45 and δ 3.66 corresponded to the protons adjacent the electronegative oxygen atom, CH2 groups (5) and (6). Although some J-values were measured, none were used to assign any sets of proton, since none were identical. A 13C NMR can be viewed in Appendix B that confirms the formation of Compound 12. 71 3,8 CDCl3 9 3 1 4 H N O 7 9 O N 2 5 6 O N H O 9 8 3 9 5,6 J = 5.90Hz J = 5.15Hz 1,2 4,7 J = 5.80Hz Figure 38. 1H NMR (500 MHz, CDCl3) of Compound 12. This spectrum may also be viewed (expanded) in Appendix A. Synthesis of the trivalent amino-amide core (13) To deprotect Compound 12, it was placed in TFA and dichloromethane (Scheme 10). When the disappearance of Compound 12 was confirmed by TLC, the reaction was quenched with water and evaporated. To further remove TFA, the sample was lyophilized several times. Compound 13 was recovered in quantitative yields and verified by NMR and mass spectrometry (Appendix D). The 1H NMR (Figure 39) for Compound 13 was easy to analyze. Now that the bulky Boc group was removed, the inner core protons peaks were well-defined and displayed clear splitting patterns. Through the coupling constants, every CH2 group 72 could be assigned, even though some values were not identical. The signal at δ 2.52 was assigned to CH2 group (3), because it was the most shielded set of protons. Since the peak at δ 3.71 was in the appropriate area and it had a J-value (5.95 Hz) close to CH2 group (3, J = 6.00 Hz), it was assigned CH2 group (4). The next set of triplets that were found to couple were CH2 group (5) at δ 3.12 (J = 5.05 Hz) and CH2 group (6) at δ 3.65 (J = 5.05 Hz). The last set triplets between δ 3.38 and δ 3.55 corresponded to CH2 (1) and CH2 (2), which have different coupling constants of 6.05 Hz and 6.25 Hz. Since CH2 groups (1) and (2) appear to have similar chemical environments, they were not assigned to one of these particular signals. Another aspect of Figure 39 that identifies the formation of Compound 13 is the proton count. Each triplet displayed 6 protons, adding up to a total of 36, which matches the expected number of protons in Compound 13. 73 1 3 H N 6 O NH3+ N 2 O 4 4, J = 5.95 Hz 5 3 5, J = 5.05 Hz 1,2 3, J = 6.00 Hz J = 6.25 Hz J = 6.05 Hz 6, J = 5.05 Hz Figure 39. 1H NMR (500 MHz, D2O) of Compound 13. This spectrum may also be viewed in Appendix A. Synthesis of maltonic acid (14) Scheme 11 outlines the reaction performed to create Compound 14. In this reaction, a three necked flask was required. Methanol and iodine were combined together in the flask and an addition funnel was attached. The reaction was heated (40ºC) before the addition of maltose, which was added to the mixture after being dissolved in a small amount of boiling water. To this concoction, a mixture of KOH and methanol was added dropwise from the addition funnel. The formation of Compound 14 could be visualized as it precipitated out of solution. After about 85 minutes, the reaction was 74 removed from the oil bath. The sample was then vacuum filtered. The precipitate was rinsed with cold methanol, then cold ethyl ether. The filtrate was subsequently filtered and washed a second time to capture the fine precipitate product. From the first filtration, 6.74 grams were collected, and from the second, 2.54 grams were obtained. After lyophilization, this desired product 14 resembled off-white sugar crystals (85% yield). Compound 14 was characterized via NMR spectroscopy. OH OH O HO HO O H' HO O HO I2, MeOH OH O KOH, 40°C HO OH Maltose HO HO H' HO Maltonic acid ( Compound 14 ) O HO OH OH K+ O- HO O 85% Yeild Scheme 11: Maltonic acid (Compound 14) was synthesized from maltose. In Figure 40, the 1H NMR spectrum for Compound 14 can be viewed. The nonreducing end anomeric hydrogen can be seen at δ 5.16, with a J-value of 3.84 Hz. The reducing end anomeric proton is absent, demonstrating the oxidation of this anomeric carbon to a carboxylic acid. The proton count coincides with the expected 13 hydrogens for this molecule that would be visible in the D2O solvent. Compound 14 was also confirmed by 13C NMR (Appendix B). 75 OH O D2O HO HO H' HO O HO OH OH OH HO O J = 3.81Hz 9.84Hz H' J = 3.84 Hz J = 9.54Hz Figure 40. 1H NMR (300 MHz, D2O) of Compound 14. This spectrum may also be found (expanded) in Appendix A. Synthesis of trivalent amino-amide glycodendrimer (15) Compound 15 was created utilizing the conditions outlined in Scheme 12 to form the amide bonds. The trivalent core 13 was placed under nitrogen gas first, followed by the addition of Compound 14. Four equivalents of Compound 14 were used in this reaction as there were 3 amine sites on Compound 13. Compounds 13 and 14 were dried further under nitrogen gas for one hour before the addition of BOP. After the solvent DMF was added to the mixture, the base DIPEA was added in excess (7 equivalents). This reaction was allowed to stir for 3 days before stopping. 76 OH H N O O + NH3+ N O HO HO OH HO OH O Compound 13 OH HO Compound 14 3 HO O BOP, DIPEA, DMF, N2 OH O HO HO OH OH O OH HO Compound 15 3.4% Yield OH O O H N N O N H 3 Scheme 12: The formation of Compound 15 through the coupling of Compound 13 and Compound 14. When the reaction was stopped it was concentrated in vacuo. To remove leftover DMF, the residue was reconstituted in MeOH and then toluene and concentrated using rotary evaporation. Next, an extraction was performed using water and CHCl3. The aqueous layer collected was lyophilized overnight before purifying by size exclusion FPLC while monitoring at 214 nm. Multiple samples were collected at this absorbance and concentrated by lyophilization. After 1H NMR analysis showed the presence of impurities, this sample was further purified by RP-HPLC to remove the remaining partially formed products and BOP byproducts. To do this, different methods were performed. A linear gradient was applied for the first trial, starting with 100% H2O, 0.0% 77 acetonitrile (ACN), and 0.10% TFA and ending with 100% ACN, 0.0% H2O. In this 60 minute run, the sample collected at 22-28 minutes demonstrated impurities, partially formed products and BOP byproducts based on 1H NMR analysis. For the next trial, a more polar method was employed. This trial started with the elution of 100% H2O, 0.10% TFA for the first 30 minutes, then a linear gradient was applied for the next 30 minutes ending with 100% ACN, 0.10% TFA. This time, the sample was collected at 5054 minutes. This sample still had the same impurities as before, so a new method was performed using a less polar conditions. The last method used started with 90% H2O, 10% ACN and ended with 70% H2O, 30% ACN after 70 minutes. A pure sample of Compound 15 (white solid) was collected in low yield (3.4%) between 13-14 minutes. The low yield was most likely due to incomplete product formation when the reaction was stopped after 3 days. It was also possible that the low yield was due to the many purification steps performed on Compound 15, causing product loss. The sample collected by HPLC was first characterized by 1H NMR spectroscopy (Figure 41). In this spectrum, there is a coupling (J = 2.35 Hz) between the doublet at δ 4.34 and the doublet of doublets at δ 4.22. These protons are part of the sugar moiety. The non-reducing sugar proton, can be seen in the insert, with a J value of 3.94 Hz. The signal at δ 2.58 was assigned to the CH2 group (5) located adjacent to the carbonyl, which shields it, moving it upfield. The other protons in Compound 15 are found overlapping, between δ 3.40 and 4.00. To integrate this spectrum, the chemical shift at δ 2.58 was calibrated for 6 protons. Since Compound 15 was evaluated in D2O, the overall proton count (75) was the same as the expected protons (75). Compound 15 was also analyzed 78 by 13C NMR (see Appendix B). The mass spectrum for Compound 15 can be found in Appendix D, further confirming this product. J = 3.95Hz H' 1 OH O HO HO H' OH OH O OH HO OH O O 3 H N 2 6 4 O 5 N H N 7 1-4,6,7 3 J = 2.35Hz 5, J = 5.90Hz J = 2.35Hz J = 6.25Hz Figure 41. 1H NMR (500 MHz, D2O) of Compound 15. This spectrum is also in Appendix A, in expanded view. Synthesis of the Boc-protected hexavalent amino core (16) Compound 16 was created using the conditions seen in Scheme 13. Compound 4 was placed under nitrogen gas then dissolved in dry MeOH. (Boc)2O was added in excess (12 equivalents) to accommodate the six reactive sites on Compound 4. The catalyst, NiCl2·6 H2O (Nickel chloride hexahydrate, 0.60 equivalents), was added next, followed by sodium borohydride (NaBH4). Since NaBH4 was added directly to the 79 reaction, it was added in portions to keep the hydrogen formation in check. After each addition, more NaBH4 was added, once the formation of bubbles had ceased. The reaction was monitored by TLC using ninhydrin stain to verify the disappearance of the starting material (Compound 4) before ending the procedure. NC Boc NH Boc O HN CN O NC O O N O N (Boc)2O, NiCl2 . 6H2O N N H N Boc O NC O Compound 4 N H N Boc N MeOH, N2 N O O O CN N Compound 16 O O NC Boc NH HN Boc TFA, CH2Cl2 NH2 H2N O O N H2N O N O NH2 N N Compound 17 O NH2 O NH2 Scheme 13: The hexavalent nitrile core 4 was reduced forming Compound 16, followed by deprotection to form an amine Compound 17. 80 After 24 hours, the reaction was stopped with the addition of tris (2aminoethyl)amine to complex with the nickel chloride hexahydrate. After 30 minutes of stirring, the reaction mixture was purple in color, confirming the complex formation. The reaction was concentrated in vacuo and then extracted two times. To analyze Compound 16, a 1H NMR was performed. Due to the long relaxation times (vide supra) of this molecule, the chemical shifts and integrations were impossible to determine. Because of this, a sample of Compound 16 was analyzed by IR (Figure 42). After verifying the disappearance of the nitrile peak (2251 cm-1) from Compound 4 and the appearance of the carbamate carbonyl peak at 1693 cm-1. Compound 16 was carried forward and used in the second step in Scheme 13. 90.0 88 86 84 1391.18 82 80 3344.25 Carbamate NH 1526.51 2865.66 2931.76 %T 1251.06 1365.79 Boc 78 N H Carbamate carbonyl O 76 N N 1172.26 O 74 1115.89 1693.84 H N Boc 72 3 70.0 3750.0 3000 2000 1500 1000 750.0 cm-1 Figure 42. IR of Compound 16 displaying a carbamate carbonyl peak at 1693.8 cm-1. 81 Synthesis of the hexavalent amino core (17) To produce Compound 17, Compound 16 was dissolved in CH2Cl2, followed by the addition of TFA (Scheme 13). The mixture was stirred at room temperature and monitored by TLC with ninhydrin staining. Compound 16 displayed a pinkish streaky band with ninhydrin stain, so when this was absent from view via TLC, the reaction was stopped. This occurred after 85 minutes. Once the reaction was stopped, it was concentrated in vacuo and then freeze dried. The sample was then dissolved in water and extracted with CHCl3. The aqueous layer was lyophilized overnight, and further purified by RP-HPLC. The samples collected between 20-70 minutes resembled formation of Compound 17 by NMR analysis, even the waste collected in between each peak. Therefore, all the samples collected in this time frame were pooled together and lyophilized. Because Compound 17 does not have a chromophore, it was thought that, since it was complexed with TFA, it would be visible through UV detection. This was not the case though; peaks that were seen were very weak. Because HPLC did not purify Compound 17, FPLC was used next. During this process, the flow rate decreased from 0.25 mL/min to ~ 0.10 mL/min and the elution of Compound 17 took four days. It is unclear at this time why this occurred. The fractions that demonstrated an absorption at 225 nm, were pooled together and concentrated by lyophilization. Compound 17 was analyzed by NMR spectroscopy and mass spectrometry. Most the peaks in the 1H NMR spectrum (Figure 43) were broad due to long relaxation times (vide supra). Even though all the chemical shifts were unclear, it was 82 still obvious where some CH2 groups should appear. Starting with the peaks furthest upfield, these relate to the most shielded protons, CH2 groups (4) and (7), since they are both adjacent methylene groups. The set of peaks between δ 2.55 and δ 3.10 corresponded to CH2 groups (1, 2, 3, and 8). The last set of peaks between δ 3.55 and δ 3.65 were the most deshielded set of protons, CH2 groups (5) and (6), which are adjacent an oxygen atom. The expected proton count for Compound 17 in a D2O solvent is 84, which is what is observed in Figure 43. The formation of Compound 17 was further confirmed through 13C NMR spectroscopy, which can be found in Appendix B. 7 4 N N 4 2 5,6 NH2 O 1 7 O 3 8 6 5 3 5 1,2,3,8 6 NH2 8 3 4,7 Figure 43. 1H NMR (500 MHz, D2O) of Compound 17 created from Scheme 13. This spectrum is also in Appendix A, in an expanded view. 83 The mass spectrum for Compound 17 can be seen in Figure 44. The peak that most correlated to Compound 17, in Figure 44 was 838.88 m/z [M+2H]. Since Compound 17 had six primary amines and four tertiary amines, it was likely that, since it was created under acidic conditions, it was harboring some acidic protons, adding to the mass. It was also possible that since this is a large molecule, it may have contained some isotopes that would increase the mass. C42H96N10O6 Exact Mass: 836.7514 NH2 O N N O NH2 3 [M+2H] Figure 44. Time of flight positive mode electrospray mass spectroscopy (TOF ES+ MS) of Compound 17. 84 Chapter 3 CONCLUSIONS AND FUTURE WORK The fight against HIV is ongoing. At least 33.3 million people are currently infected worldwide.1,2 There are treatments for HIV, but they are not without drawbacks, like viral resistance and harmful side-effects.1 Most drug treatments thus far attack HIV from within the cell. There are few drugs that prevent HIV from binding to the host cell and inhibit infectivity at a very early stage in the process. It has been established that sulfated molecules have an affinity to HIV through ionic interactions, between the viral surface gp120 and the polyanionic sulfated compounds. Some studies have shown that sulfated glycodendrimers have an affinity to HIV through these interactions and thus have the potential to prevent HIV infection.5,6,39 As sulfated glycodendrimers have shown the potential to inhibit HIV infection, the synthesis of this class of molecules is of profound interest in the fight against HIV. In this research, two glycodendrimers, 10 and 14, were created. A hexavalent amino core 17 was also created, which will be utilized later to synthesize other glycodendrimers. Creating these glycodendrimers is only one part of process. To further prepare these molecules for the testing of their HIV inhibition potential, both glycodendrimers need to be sulfated. As mentioned before (vide supra), Compound 10 has already been sulfated (Scheme 7) by another group member.50 Eventually this process will be performed on Compound 14 as well. 85 After Compounds 10 and 14 are sulfated, they will be tested with an ELISA (enzyme-linked immunosorbent assay). This is a quick competitive gp120 binding assay that will be utilized to screen the glycodendrimers for binding affinity.5 If Compounds 10 and 14 demonstrate binding affinity, the samples will be tested on active viral particles by a collaborator at Duke University. At Duke University, a luciferase reporter gene assay will be used to determine how well Compounds 10 and/or 14 are able to inhibit HIV-1 infection.5 If Compounds 10 and 14 demonstrate inhibitory properties, it may lead to the development of new anti-viral agents to fight HIV infection. These glycodendrimers may be used as microbicides, preventing the spread of HIV. While it is important to find treatments for HIV patients, it is also important to help find ways to protect individuals not infected with the virus. As the old saying goes, “An ounce of prevention is worth a pound of cure”.51 86 Chapter 4 EXPERIMENTAL Materials Dichloromethane (CH2Cl2), tris(2-aminoethyl)amine, potassium carbonate (K2CO3), acetyl chloride and p-toluenesulfonyl chloride (TsCl), Di-tert-butyl dicarbonate ((Boc)2O), N,N-diisopropylethylamine (DIPEA), and dimethyl sulfoxide (DMSO), were purchased from Acros Organics, and sodium hydroxide (NaOH) and potassium hydroxide (KOH) from Spectrum Chemicals. Ethyl acetate (EtOAc), hexane, and silica gel were purchased from Whatman. Maltose and 1,3-propanediol were obtained from Sigma-Aldrich, and methanol (MeOH) and lithium hydroxide (LiOH) from Fisher Scientific. Nickel chloride (NiCl2·6H2O) was purchased from Alfa-Aesar. Anhydrous MeOH, dichloromethane (CH2Cl2), and acetonitrile (ACN), and sodium borohydride (NaBH4) were obtained from EMSci. Trifluroacetic acid (TFA), triethylamine (TEA), ammonium bicarbonate (NH4HCO3), dimethylformamide (DMF) were purchased from EMD and O-(benzotriazol-1-yl)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) from Chem-Impex International. Benzotriazol-1-yloxy-tris(dimethylamino) phosphonium hexafluorophosphate (BOP) was obtained from Advanced Chemical Technology. The 100, 500, and 2000 MW cellulose ester membrane were purchased from Spectra/Por®, and the P-10 Bio-Gel used was from Bio-Rad. The materials used in this research were not purified prior to use, and only when reactions conditions involved the use of nitrogen gas were all glassware, syringes, stir-bars, etc., flame or oven-dried. 87 Instrumentation A Bruker Avance 300 or Avance III 500 NMR (NSF, CHE MRI-0922676) were used for all NMR measurements. All lyophilization was performed using the Freeze Dry/Shell Freeze System (LABCONCO 7522800). A 2032 DryFast Ultra® was used for all rotary evaporation. All centrifugation was performed using Centrific Model 228 (Fisher Scientific). A Hewlett Packard TI-series 1050 with a Grace Prevail C-18 5µM column (10 x 250 mm) were used for the reverse phase high pressure liquid chromatography (RP-HPLC). Fast pace liquid chromatography (FPLC) was performed on two different pump systems; one was a Pharmacia pump P-500 with a L.C. Controller LCC-500 Plus, and the other was a Bio-Rad BioLogic DuoFlow F10 Pumphead with a Dell Controller. A Bio-Rad Econo-Column (2.5 x 120 cm) was used with both FPLC systems. The microwave synthesis was performed with a microwave accelerated reaction system (MARS®). A Perkin Elmer System 2000 FT-IR Spectrometer was used for all IR measurements. Characterization The 1H and 13C spectra were analyzed using Spinworks or Topspin. Solvents used were either deuterium oxide (D2O) or chloroform-d (CDCl3) and purchased from Acros. Expanded versions of all spectra can be located in the Appendices; 1H NMR in Appendix A, 13C NMR in Appendix B, infrared (IR) in Appendix C and mass spectra in Appendix D. The electrospray time-of-flight (ES-TOF) were collected from Ohio State 88 University, Mass Spectrometry & Proteomics Facility. The MALDI-MS were obtained from Ohio State University, Mass Spectrometry & Proteomics Facility, and the University of the Pacific Mass Spectrometry Facility. Methods Synthesis of 3-(3-hydroxypropoxy)propanenitrile (1)40 Using a syringe, 62.5 μl of 40% NaOH (W/V) was added dropwise to 1,3propandiol (0.95 mL, 13.1mmol) in a round bottom flask (25 mL). The mixture was placed in a water bath upon the addition of acrylonitrile (0.39 mL, 5.88 mmol), which was added dropwise. TLC (90% EtOAc, 10% hexane, with visualization using I2 and molybdenum stain) was performed to verify the disappearance of the starting material, before stopping the reaction. The procedure was allowed to run 3 days, and then quenched with DI H2O. Next it was acidified to ~ pH 7 with 1 M HCl. The residue was next concentrated by freeze drying and purified by flash silica gel chromatography (90% EtOAc, 10% hexane) and isolated by rotary evaporation. Compound 1 (clear pale yellow liquid) was collected at Rf = 0.36. From the reaction, 559 mg (74% yield) of Compound 1 was obtained. 1H NMR (500 MHz, CDCl3): δ1.70 (app. pentet, 2H, J = 6.10Hz), 2.50 (t, 2H, J = 6.20 Hz), 2.93 (s, 1H), 3.50 (t, 2H, J = 6.05Hz), 3.52 (t, 2H, J = 6.25Hz), 3.58 (t, 2H, J = 6.05Hz). 68.6, and 118. C NMR (300 MHz, CDCl3): δ 18.5, 31.8, 59.6, 65.1, 13 89 Synthesis of 3-(2-cyanoethoxy)propyl p-toluenesulfonate (3)42 The starting material (1, 1.0 g, 7.9 mmol) was dissolved in dichloromethane (13 mL) and heated to 50-54ºC in a round bottom flask (50 mL). Tosyl chloride (3.00 g, 15.8 mmol) was added to the mixture, followed by the dropwise addition 2.9 mL of 50% NaOH (W/V) dropwise. The reaction was monitored using TLC (50% EtOAc 50% hexane and UV, molybdenum stain) and after the disappearance (6 days) of Compound 1, the reaction was allowed to reach room temperature. An extraction was performed with water (3x), followed by rotary evaporation of the organic phase. Next, the product was purified through silica gel flash chromatography (50% EtOAc and 50% hexane) and collected at the Rf value of 0.39. Compound 3 (679 mg, 30.4% yield, viscous cloudy white liquid) was characterized by NMR spectroscopy. 1H NMR (500 MHz, CDCl3): δ1.92 (m, 2H, J = 5.95Hz), 2.43 (s, 3H), 2.50 (t, 2H, J = 6.35Hz), 3.50 (t, 2H, J = 5.95Hz), 3.54 (t, 2H, J = 6.35Hz), 4.12 (t, 2H, J = 6.00Hz), 7.34 (d, 2H, J = 7.95Hz), 7.7 (d, 2H, J = 8.25Hz). C NMR (300 MHz, CDCl3): δ 18.4, 21.3, 28.8, 65.1, 66.2, 67.2, 13 118, 128, 130, 133, and 145. Synthesis of hexavalent nitrile core (4)42 In a round bottom flask (100mL) with a condenser attached, anhydrous acetonitrile (30.0 mL) and K2CO3 (2.83 g, 20.5 mmol) were mixed under N2. Upon the addition of the tris(2-aminoethyl)amine (0.40 mL, 2.67 mmol) the reaction was heated to 89ºC. Next, a mixture of the Compound 3 (5.80 g, 20.5 mmol), and dry acetonitrile (2 90 mL) were added in portions over a four hour period and the temperature was adjusted to 85ºC. Over the next 20 hours, the temperature was lowered to 82ºC and allowed to reflux at this temperature until the reaction was stopped. Silica gel TLC (CHCl3 : MeOH : triethylamine (1:1:0.1%), I2, ninhydrin stain) was performed to verify the disappearance of the starting material (tris-core). On the second day when the yellowish color verifying the presence of the tris-core on the TLC plate was missing, the reaction was stopped. After the reaction cooled to room temperature, it was filtered, through filter paper and concentrated in vacuo. The residue (bronze colored oil) was then extracted using H2O (200mL) and CHCl3 (3x 75 mL). Next, the organic layer was concentrated in vacuo and then washed in water followed by centrifugation (3400 rpm) for 10 minutes. The supernatant was decanted into a round bottom flask (100 mL) containing an anion exchange resin (AG 1-X8, 50-500 mesh, chloride form) and stirred overnight, then filtered and lyophilized. This process was repeated with the pellet collected to obtain any remnants of the desired product 4 not collected in the first water wash. From the combined samples, a dark bronze viscous liquid was obtained for Compound 4 (2.03g, 91% yield). 1H NMR (300 MHz, CDCl3): δ 1.66 (m, 12H), 2.46 (m, 24H), 2.56 (t, 12H, J = 6.21Hz), 3.48 (t, 12H, J = 6.27Hz), 3.59 (t, 12H, J = 6.27Hz); 13C NMR (500 MHz, CDCl3): δ 18.5, 27.0, 50.7, 52.1, 53.0, 65.0, 69.0, 118. IR (CHCl3): 665.8, 755.1, 844.4, 951.8, 1118, 1225, 1328, 1368, 1414, 1467, 1668, 2251, 2873, and 3018 cm-1. MALDITOF MS [M + H] (C42H72N10O6) Calcd: m/z = 813.6, Found: m/z = 813.8. 91 Synthesis of methyl ester core (5)46,52 Compound 4 (504 mg, .619 mmol) and anhydrous MeOH (2.0 mL) were combined in a round bottom flask (10 mL) and stirred in an ice bath. After the acetyl chloride (1.6 mL) was added, the reaction was stirred overnight at room temperature. TLC (CHCl3 : MeOH : H2O (6:4:0.5), I2, ninhydrin) was performed to verify the disappearance of the starting material by colorimetry. After 24 hours, the brownish color representing Compound 4 had disappeared and the reaction was stopped. It was evaporated, and freeze dried overnight, and then dialyzed for 2 hours in 100 MWCO tubing against 4L of water. A 217 mg (33% yield) sample (off-white chunky solid) was recovered after lyophilization. After Compound 5 was characterized, it was carried forward in the following reaction to create a carboxylic acid terminated core. IR (acetone): 1113, 1197, 1733, and 2924 cm-1. Synthesis of the hexavalent carboxy core (5)47 Compound 5 (109 mg, 0.10 mmol) was transferred to a round bottom flask (25 mL) and dissolved into nanopure water (1.5 mL). After LiOH (60 mg, 2.5 mmol) was added to the mixture, it was heated to 60ºC in a microwave (400 W) for 5 minutes before the addition of more H2O (1.0 mL). The reaction was reheated to 60ºC for 20 more minutes in the microwave. When the sample reached room temperature, it was concentrated in vacuo, washed with MeOH (2 mL) and centrifuged (3400 rpm) for 10 minutes. The supernatant was decanted and concentrated in vacuo and further purified 92 using size exclusion FPLC (2.5cm x 120cm column, Bio-Rad P-10 size exclusion gel). The column was eluted with 0.03M ammonium bicarbonate at a rate of 0.25mL/min and monitored at 214 nm. The peaks collected resulted in 66% yield (68.1 mg) of Compound 6 (tannish colored chunky solid). 1H NMR (500 MHz, D2O): δ 1.85 (m, 12H), 2.45 (t, 12H, J = 6.60Hz), 2.75-2.91 (m, 24H), 3.58 (t, 12H, J = 6.10Hz), 3.71 (t, 12H, J = 6.60 Hz); 13C NMR (500 MHz, D2O, internal MeOH reference): δ 26.0, 37.8, 49.4, 50.2, 50.8, 67.8, 68.5, 180. ES TOF MS [M - H] (C42H78N4O18) Calcd: m/z = 925.5233, Found: m/z = 925.5223. Synthesis of the Boc-protected oxime sugar-linker (8)53 Compound 7 (125 mg, 0.54 mmol) and maltose (222 mg, 0.62 mmol) were combined in a round bottom flask (25 mL). Ammonium acetate (0.1M, 5 mL, pH 4.5) was added next. After the mixture stirred for 30 minutes, the pH was checked. The pH was found to be 4.5 and the reaction was allowed stir overnight. Before stopping the reaction after 20 hours, the pH was checked again. Throughout the reaction the pH remained around 4.5. TLC (CHCl3:MeOH:H2O, 6:4:0.5, with visualization using molybdenum stain) was performed to verify the disappearance of Compound 7 (Rf = 0.80) before stopping the reaction. After the reaction was complete, it was freeze dried. The residue was then purified with flash silica gel chromatography using the same TLC conditions as described above. From the flash column, Compound 8 (Rf = 0.53, 231 mg, 76% yield, white crystalline solid) was recovered and analyzed by NMR spectroscopy. 1 H NMR (500 MHz, D2O): δ 1.45, (s, 9H), 1.78 (t, 2H, J = 6.60Hz), 3.14 (t, 2H, J = 93 6.60Hz) 3.48 (m, 1H), 3.60-4.00 (m, 14H), 4.12 (m, 0.2H), 4.35 (m, 2H), 4.60 (m, 1H), 5.00 (m, 0.2H), 5.12 (d, 1Hα, J = 3.90Hz), 5.40 (m, 0.1H), 6.98 (d, 0.1HZ, J = 5.50Hz) and 7.60 (d, 0.6HE, J = 6.05Hz); 13C NMR (500 MHz, D2O, internal MeOH reference): δ 16.9, 27.8, 28.7, 28.8, 37.1, 48.9, 57.4, 60.4, 60.5, 60.8, 62.1, 62.3, 65.7, 68.3, 68.4, 68.5, 68.7, 71.3, 71.7, 71.8, 72.2, 72.3, 72.5, 72.7, 72.9, 73.0, 73.1, 73.4, 75.8, 76.9, 77.2, 80.2, 81.5, 90.2, 99.8, 100, 101, 152, 153, 158. Synthesis of the E,Z oxime sugar-linker (9)54 Dichloromethane (3 mL) and Compound 8 (231 mg) were combined in a round bottom flask (25 mL). Next, TFA (1.5 mL) was added dropwise over a 16 minute period. The reaction was stirred at room temperature and monitored by TLC (6:4:1 CHCl3:MeOH:H2O, ninhydrin stain). When the starting material (Rf = 0.64, light pink color) was absent from view by TLC and replaced by a new product (Rf = 0.42, streaky pink purplish color), the reaction was stopped (3 hours). The reaction was quenched with water (0.5 mL) and then concentrated in vacuo. The residue was dissolved in ammonium bicarbonate and then lyophilized. This was repeated one additional time. The desired product 9 (white crystalline solid) was produced in quantitative yield. 1H NMR (500 MHz, D2O): δ 1.94 (m, 2H), 3.08 (m, 2H), 3.40 (m, 1H), 3.50-4.00 (m, 15H), 4.10 (m, 0.1H), 4.30 (m, 2H), 4.50 (t, 0.4H, J = 5.15 Hz), 4.95 (m, 0.1H), 5.12 (d, 0.5Hα, J = 3.90 Hz), 5.40 (d, 0.4H, J = 3.95 Hz), 6.98 (d, 0.09HZ, J = 5.55 Hz) and 7.60 (d, 0.4HE, J = 5.95 Hz); 13C NMR (500 MHz, D2O, internal MeOH reference): δ 26.5, 26.6, 37.6, 37.7, 37.7, 37.8, 60.5, 60.6, 60.7, 60.8, 60.9, 62.2, 62.4, 65.7, 68.0, 68.4, 68.5, 68.8, 69.0, 69.3, 94 69.4, 70.0, 71.3, 71.4, 71.7, 71.8, 71.9, 72.1, 72.6, 72.7, 72.9, 73.0, 73.1, 73.2, 73.3, 74.1, 74.6, 75.8, 76.2, 76.9, 77.0, 77.2, 80.2, 81.2, 90.0, 91.9, 95.8, 100, 152, 153. ES TOF MS [M+Na] (C17H34N2O12) Calcd: m/z = 481.2009, Found: m/z = 481.1995. Synthesis of hexavalent maltose amino-oxime glycodendrimer (10)49 Compound 9 (331 mg, 0.578 mmol) and Compound 10 (84.8 mg, 0.0916 mmol) were added together to a round bottom flask (50 mL) and dissolved in nanopure water (2mL). The mixture was freeze dried overnight. The lyophilized sample was placed under N2 gas and dissolved in DMSO (2 mL). The addition of TBTU (424 mg, 1.32 mmol) was next, followed by DIPEA (300 μL). DIPEA was added in portions (100 μL), to a pH of 9. When the reaction was complete after 24 hours, it was lyophilized. Next, an extraction was performed. The residue was dissolved in water (50 mL) and impurities were extracted 3x with CHCl3 (20mL). After the aqueous phase was lyophilized, the residue was dissolved back into water and any insoluble products were filtered through glass wool. Dialysis was performed next using 2000 MWCO tubing for a total of 7.5 hours at 4ºC, with water changes at least every hour. After the sample was freeze dried again, it was filtered through glass wool to remove any ppt. The sample recovered (215 mg, 66% yield) had the appearance of light brown sugar. In the 1H NMR only one arm was integrated (500 MHz, D2O): δ 1.79-2.00 (m, 4H), 2.59 (m, 2H), 2.70-3.20 (m, 4H), 3.30 (m, 2H), 3.45 (m, 2H), 3.50-4.01 (m, 19H), 4.26 (m, 2H), 4.54 (m, 0.4H), 5.00 (m, 0.1H) 5.12 (d, 0.6H, J = 3.70 Hz), 5.40 (m, 0.3H), 7.00 (d, 0.1HZ, J = 5.40 Hz), and 7.60 (d, 0.5HE, J = 5.95 Hz). 13C NMR (500 MHz, D2O, internal MeOH reference): δ 24.5, 95 28.6, 35.9, 36.6, 39.2, 39.6, 47.6, 50.3, 50.4, 51.2, 51.3, 51.4, 60.5, 60.6, 60.7, 60.9, 61.0, 62.2, 62.5, 65.8, 66.8, 67.6, 68.4, 68.5, 68.6, 68.7, 68.9, 69.4, 69.5, 71.4, 71.8, 72.3, 72.4, 72.6, 72.8, 72.9, 73.0, 73.1, 73.2, 73.5, 75.9, 76.9, 77.3, 80.4, 81.6, 90.3, 99.8, 100, 101, 152, 154, 174. MADLI-TOF MS, [M+H] (C144H270N16O84) Calcd: m/z = 3568.735, Found: m/z = 3570.280. Synthesis of the Boc-protected trivalent amino-amide core (12)55 Tris(2-aminoethyl)amine (51.0 μL, 0.342 mmol) was added to recently freeze dried Compound 11 (290 mg, 1.25 mmol) in a round bottom flask (25 mL). The sample was immediately placed under nitrogen gas. BOP (605 mg, 1.37 mmol) was added next, followed by anhydrous DMF (1 mL). After the mixture was stirred for 5 minutes, DIPEA (400 μL, 2.39 mmol) was added. The reaction was allowed to stir for three days before stopping. After the sample was concentrated by rotary evaporation, an extraction was performed. The residue was dissolved in nanopure water (40 mL) and extracted 3x with chloroform (20 mL). The organic layer was then extracted 3x with water (20 mL). The organic phase was concentrated in vacuo before purifying by flash silica gel chromatography (3x). With the first flash column CHCl3:MeOH (8:1 and ninhydrin stain) were employed to elute Compound 12 (Rf = 0.48). On the second flash column, the product was eluted with ethyl acetate (100%), then ethyl acetate:methanol (95:5), followed by methanol (100%). Compound 12 was visible eluting during the addition of 100% methanol and collected. For the last column, a silica plug 4 cm in height was utilized and the same solvent conditions as column two. When Compound 12 was 96 visible eluting during methanol addition, it was collected and concentrated in vacuo. The sample (72.3 mg, 27% yield) characterized by NMR. 1H NMR (500 MHz, CDCl3): δ 1.42 (s, 27H), δ 2.43 (t, 6H, J = 5.80 Hz), 2.52 (m, 6H), 3.22 (m, 12H), 3.45 (t, 6H, J = 5.15 Hz), 3.66 (t, 6H, J = 5.90 Hz), 5.30 (s, 3H), 7.05 (s, 2H). 13 C NMR (500 MHz, CDCl3): δ 29.4, 37.6, 41.2, 55.3, 70.8, 80.1, 157, 173. Synthesis of the trivalent amino-amide core (13)54 Compound 12 (72.3 mg, 0.0913 mmol) and CH2Cl2 were added together in a round bottom flask (10 mL). TFA (500 μL, 50% (v/v)) was added next dropwise. TLC (methanol, 100%) was performed to verify the disappearance of Compound 12. The reaction was stopped and quenched with water (0.5 mL) after 1 hour. The residue was concentrated in vacuo. The sample was then lyophilized (2x) overnight. Compound 12 (pale rust colored sticky solid) was characterized by 1H NMR (500MHz, D2O): δ 2.52 (t, 6H, J = 6.00 Hz), 3.12 (t, 6H, J = 5.00 Hz), 3.38 (t, 6H, J = 6.25 Hz), 3.55 (t, 6H, J = 6.25 Hz), 3.66 (t, 6H, J = 5.05 Hz), 3.72 (t, 6H, J = 6.00 Hz). 13 C NMR (500 MHz, D2O, internal MeOH reference): δ 34.5, 35.7, 39.1, 53.4, 66.3, 66.6, 175. ES-TOF MS [M+H] (C21H45N7O6) Calcd: m/z = 492.3509, Found: m/z = 492.3500. Synthesis of maltonic acid (14)56 All glassware used in this experiment was rinsed with methanol prior to use. Methanol (238 mL) and iodine (13.4 g, 52.7 mmol) were combined together in a threeneck round bottom flask (1 L) and heated to 40ºC. Maltose (10.0 g, 27.8 mmol) was 97 dissolved in boiling water (~20 mL) before adding to the mixture of methanol/iodine. Next, a mixture of KOH (14.3 g, 254 mmol) and MeOH (357 mL) were mixed together and then added dropwise to the reaction via an addition funnel. After 85 minutes, the reaction was removed from the oil bath and allowed to stir at room temperature for 95 minutes. The residue was filtered and rinsed with cold MeOH (1400 mL) followed by cold ethyl ether (1000 mL). To collect product lost through the filtration, the eluent collected was filtered again, this time by gravity through 4 layers of filter paper, and rinsed with cold ethyl ether (500 mL). After lyophilization, the residue (off-white crystals, 9.28 g, 85% yield) was characterized through NMR spectroscopy. 1H NMR (300 MHz, D2O): 3.14 (t, 1H, J = 9.54Hz), 3.51-3.55 (dd, 1H, J = 3.81Hz, 9.54Hz), 3.643.98 (m, 9H), 4.07-4.12 (m, 2H), 5.14 (d, 1H, J = 3.84Hz). 13 C NMR (300 MHz, D2O, internal MeOH reference): δ 60.4, 62.2, 69.4, 71.8, 72.4, 72.6, 72.7, 73.0, 82.4, 100, 178. Synthesis of the trivalent maltose amino-amide glycodendrimer (15)55 Compound 13 (83.7 mg, 0.100 mmol) and Compound 14 (168 mg, 0.424 mmol) were added together in a round bottom flask (50 mL) and placed under nitrogen gas. After 1 hour, BOP (199 mg, 0.450 mmol) was added to the mixture followed by DMF (5 mL) and DIPEA (70.0 μL, 0.418 mmol). The reaction was stirred at room temperature for 3 days before stopping by the addition of MeOH (20 mL). Once the sample was concentrated in vacuo, toluene (20 mL) was added next, followed by rotary evaporation. The sample was reconstituted in water (50 mL) and extracted with 20 mL CHCl3 (4x). 98 The aqueous phase was freeze dried overnight and then purified via FPLC (2.5cm x 120cm column, Bio-Rad P-10 size exclusion gel). The column was eluted with 0.03M ammonium bicarbonate at a rate of 0.25mL/min while monitoring at 214 nm. The peaks collected were concentrated by lyophilization. The sample was further purified by RPHPLC. This procedure was performed multiple times with different gradients. The only constant value in each method was the addition of 0.10% TFA (v/v) in each trial. The first trial started with a linear gradient with 100% H2O, 0.0% ACN and ended with 100% ACN, 0.0% H2O after 60 minutes. The sample collected between 22 and 28 minutes was concentrated by lyophilization. For the next trial, in the first 30 minutes 100% H2O was maintained, then a linear gradient was applied for the next 30 minutes ending with 100% ACN, 0.0% H2O. This time the sample was collected at 54 minutes. After lyophilization, the sample was purified again through RP-HPLC. The next method started with 90% H2O, 10% ACN and ended with 70% H2O, 30% ACN after 70 minutes. A pure sample of Compound 15 (5.2 mg, 3.4% yield) eluted between 13-14 minutes. After the sample was freeze dried, the residue (white sticky solid) was characterized by NMR spectroscopy. 1H NMR (500 MHz, D2O): δ 2.59 (t, 6H, J = 5.90 Hz), 3.45-3.47 (m, 14H), 3.63-4.00 (m, 46H), 4.21-4.23 (dd, 3H, J = 2.35 Hz, 6.25 Hz), 4.34 (d, 3H, J = 2.35 Hz), 5.12 (d, 3H, J = 3.95 Hz). 13C NMR (500 MHz, D2O, internal MeOH reference): δ 34.7, 35.9, 38.7, 53.8, 60.5, 62.3, 66.4, 66.8, 69.5, 71.8, 71.9, 72.0, 72.4, 72.6, 73.0, 82.1, 101, 174, 175. MALDI-TOF, [M+Na+H] (C57H105N7O39) Calcd: m/z = 1535.645, Found: m/z = 1535.159. 99 Synthesis of the Boc-protected hexavalent amino core (16)57 Compound 4 (65.4 mg, 0.0805 mmol) was placed in a flame dried round bottom flask (25 mL) and placed under nitrogen gas. Compound 4 was dissolved in MeOH (5 mL) followed by the addition of (Boc)2O ( 211 mg, 0.966 mmol) and NiCl2·6H2O (11.5 mg, 0.0483 mmol). Next, NaBH4 (128 mg, 3.38 mmol) was added to the reaction in portions over a 25 minute period. Silica gel TLC (CHCl3 : MeOH : H2O (6:4:0.5), ninhydrin staining) was performed to verify the disappearance of the starting material (Compound 4) before ending the procedure. After 24 hours, the yellow/brownish streaky band verifying the presence of Compound 4 had disappeared, and the reaction was stopped. A new pinkish band was present on the TLC plate, representing new product formation. Tris (2-aminoethyl)amine (72.3 μL, 0.483 mmol) was added to the sample and stirred for 30 minutes. An extraction was performed after the sample was concentrated in vacuo. For this procedure, the sample was dissolved in ethyl acetate (50 mL), and impurities were extracted with 20 mL of sodium bicarbonate (NaHCO3 x3). The organic layer was concentrated in vacuo, and then reconstituted in CHCl3 (20 mL). Impurities were extracted with H2O (10 mL x2). After evaporation, a sample of Compound 16 (vicous light brownish liquid) was dissolved in CHCl3 and analyzed by IR (CHCl3): 1116, 1172, 1251, 1366, 1391, 1526, 1694, 2866, 2932, and 3344 cm-1. This sample (60.2 mg, 54% yield) was carried forward without further purification. 100 Synthesis of the hexavalent amino core (17)54 Compound 16 (60.2 mg, 0.0435 mmol) and CH2Cl2 (2 mL) were added together in a round bottom flask (10 mL). TFA (1 mL, 50% (v/v)) was added next dropwise. TLC (CHCl3:MeOH:H2O, 6:4:0.5, ninhydrin stain) was performed to verify the disappearance of Compound 16. The reaction was stopped after 85 minutes and concentrated in vacuo and then the sample was freeze dried. After this, the sample was dissolved in water (20 mL) and extracted with 10 mL of CHCl3 (3x). The aqueous layer was then lyophilized overnight and purified further with RP-HPLC. The linear gradient used here started with 90% H2O: 10% ACN: 0.10% TFA and ended with 70% H2O: 30% ACN: 0.10% TFA after 70 minutes. The sample collected between 20-70 minutes was lyophilized and purified by FPLC (2.5cm x 120cm column, Bio-Rad P-10 size exclusion gel). The column was eluted with 0.03M ammonium bicarbonate at a rate of 0.25mL/min and monitored at 214 nm. The peaks collected were concentrated by lyophilization. When this product eluted the flow rate decreased from 0.25mL/min to 0.10mL/min. and it took 4 days to elute. To visualize the peaks the y-axis had to be expanded to see the product eluting, since Compound 17 has no chromophore. The desired product 17 (5.4 mg, 16% yield) collected was freeze dried and characterized first by NMR. 1H NMR (500MHz, D2O): δ 1.70-1.95 (m, 24H), 2.55-2.75 (m, 24H), 2.95-3.15 (m, 12H), 3.55-3.65 (m, 24H). 13 C NMR (500 MHz, D2O, internal MeOH reference): δ 25.5, 25.6, 27.9, 28.1, 29.7, 37.6, 38.5, 49.1, 50.2, 50.5, 50.6, 50.7, 51.0, 68.0, 68.1, 68.6, 68.7, 68.9, 69.1, 69.2. ES TOF MS [M+2H] (C42H96N10O6) Calcd: m/z = 838.77, Found: m/z = 838.88. 101 APPENDICES 102 APPENDIX A 1 H Spectra 103 3-(3-Hydroxypropoxy)propanenitrile (Compound 1), 1H NMR, 500 MHz, CDCl3. 104 3-(2-Cyanoethoxy)propyl p-toluenesulfonate (Compound 3), 1H NMR, 500 MHz, CDCl3. 105 Hexavalent nitrile core (Compound 4), 1H NMR, 300 MHz, CDCl3. 106 Hexavalent carboxy core (Compound 6), 1H NMR, 500 MHz, D2O. 107 Boc-protected oxime sugar-linker (Compound 8), 1H NMR, 500 MHz, D2O. 108 E,Z oxime sugar-linker (Compound 9), 1H NMR, 500 MHz, D2O. 109 Hexavalent maltose amino-oxime glycodendrimer (Compound 10), 1H NMR, 500 MHz, D2O. 110 Boc-protected trivalent amino-amide core (Compound 12), 1H NMR, 500 MHz, CDCl3. 111 Trivalent amino-amide core (Compound 13), 1H NMR, 500 MHz, D2O. 112 Maltonic acid (Compound 14), 1H NMR, 300 MHz, D2O. 113 Trivalent maltose amine-amide glycodendrimer (Compound 15), 1H NMR, 500 MHz, D2O. 114 Hexavalent amino core (Compound 17), 1H NMR, 500 MHz, D2O. 115 APPENDIX B 13 C Spectra 116 3-(3-Hydroxypropoxy)propanenitrile (Compound 1), 13C NMR, 300 MHz, CDCl3. 117 3-(2-Cyanoethoxy)propyl p-toluenesulfonate (Compound 3), 13C NMR, 300 MHz, CDCl3. 118 Hexavalent nitrile core (Compound 4), 13C NMR, 500 MHz, CDCl3. 119 Hexavalent carboxy core (Compound 6), 13C NMR, 500 MHz, D2O. 120 Boc-protected oxime sugar-linker (Compound 8), 13C NMR, 500 MHz, D2O. 121 E,Z oxime sugar-linker (Compound 9), 13C NMR, 500 MHz, D2O. 122 Hexavalent maltose amino-oxime glycodendrimer (Compound 10), 13C NMR, 500 MHz, D2O. 123 Boc-protected trivalent amino-amide core (Compound 12), 13C NMR, 500 MHz, CDCl3. 124 Trivalent amino-amide core (Compound 13), 13C NMR, 500 MHz, D2O. 125 Maltonic acid (Compound 14), 13C NMR, 300 MHZ, D2O 126 Trivalent maltose amino-amide glycodendrimer (Compound 14), 13C NMR, 500 MHz, D2O. 127 Hexavalent amino core (Compound 17), 13C NMR, 500 MHz, D2O. 128 APPENDIX C Infrared Spectra 129 Hexavalent nitrile core (Compound 4), IR with CHCl3. 130 Hexavalent methyl ester (Compound 5), IR with acetone. 131 Boc-protected hexavalent amino core (Compound 15), IR with CHCl3. 132 APPENDIX D Mass Spectra 133 Hexavalent nitrile core (Compound 4), MALDI TOF MS. 134 Hexavalent carboxy core (Compound 6), ESI TOF MS. 135 E,Z oxime sugar-linker (Compound 9), ESI TOF MS. 136 Hexavalent maltose oxime-amino glycodendrimer (Compound 10), MALDI TOF. 137 Trivalent amino-amide core (Compound 13), ESI TOF MS. 138 Trivalent maltose amino-amide glycodendrimer (Compound 15), MALDI TOF MS. 139 Hexavalent amino core (Compound 17), ESI TOF MS. 140 REFERENCES [1] National Institute of Allergy and Infectious Diseases, National Institutes of Health, HIV/AIDS, http://www.niaid.nih.gov/topics/HIVAIDS/Understanding/Pages/ quickFacts.aspx. Access date: 10/3/2011. Last updated: 6/2011. [2] National Institute of Allergy and Infectious Diseases, National Institutes of Health, HIV/AIDS, http://www.niaid.nih.gov/topics/HIVAIDS/Understanding/Treatment. Access date: 10/3/2011. Last updated: 6/2011. [3] US Department of Health and Human Services, http://aidsinfo.nih.gov/DrugsNew/, Access date: 10/3/2011. Last updated: 9/2011. [4] Johnson, M., Stallworth, T., Neilands, T.; The Drugs or the Disease? Causal Attributions of Symptoms Held by HIV-Positive Adults on HAART. AIDS and Behavior, 2003, 7, 109-117. [5] Clayton,R., Hardman,J., LaBranche,C.C., McReynolds, K.D.; Evaluation of the Synthesis of Sialic Acid-PAMAM Glycodendrimers without the Use of Sugar Protecting Groups, and the Anti-HIV-1 Properties of These Compounds. Bioconjugate Chemistry, 2011, 22, 2186-2197. [6] Price,C.F., Tyssen, D., Sonza, S., Davie, A., Evans, S., Lewis, G.R., Xia, S., Spelman, T., Hodsman, P., Moench, T.R., Humberstone, A., Paull, J.R.A., Tachedjian, G.; SPL7013 Gel (VivaGel®) Retains Potent HIV-1 and HSV-2 Inhibitory Activity following Vaginal Administration in Humans. PLoS ONE, 2011, 69, e24095. [7] Tyssen, D., Henderson, S., Johnson, A., Sterjovski, J., Moore, K., La, J., Zanin, M., Sonza, S., Karellas, P., Giannis, M., Krippner, G., Wesselingh, S., McCarthy, T., Gorry, P., Ramsland, P., Cone, R., Paull, J., Lewis, G., Tachedjian, G.; Structure Activity Relationship of Dendrimer Microbicides with Dual Action Antiviral Activity. PLoS ONE 2010, 5, e12309. [8] Turner, B.G., Summers, M.F.; Structural Biology of HIV. Journal of Molecular Biology, 1999, 285, 1-32. [9] Goto, T., Nakai, M., Ikuta, K.; The Life-Cycle of Human Immunodeficiency Virus Type. Micron, 1998, 29, 123-138. [10] Chan, D. C., Fass, D., Berger, J.M., Kim, P.S.; Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell, 1997, 89, 263-273. [11] Doms, R. W.; Beyond Receptor Expression: The Influence of Receptor Conformation, Density, and Affinity in HIV-1 Infection. Virology, 2000, 276, 229-237. [12] McReynolds, K. & Gervay-Hague, J.; Chemotherapeutic Interventions Targeting HIV Interactions with Host-Associated Carbohydrates. Chemical Reviews, 2007, 107, 1533-1552. [13] Moore, J.P., McKeating, J.A., Huang, Y., Ashkenazi, A., Ho, D.D.; Virions of Primary Human Immunodeficiency Virus Type 1 Isolates Resistant to Soluble CD4 (sCD4) Neutralization Differ in sCD4 Binding and Glycoprotein gpl20 Retention from sCD4-Sensitive Isolates. Journal of Virology, 1992, 66, 235-243. [14] Sierra, S., Kupfer, B., Kaiser, R., Basics of the Virology of HIV-1 and Its Replication. Journal of Clinical Virology, 2005, 34, 233-244. 141 [15] de Parseval, A., Bobardt, M. D., Chatterji, A., Chatterji, U., Elder, J. H., David, G., Zolla-Pazner, S., Farzan, M., Lee, T.-H., Gallay, P.A.; A Highly Conserved Arginine in gp120 Governs HIV-1 Binding to Both Syndecans and CCR5 via Sulfated Motifs. The Journal of Biological Chemistry, 2005, 280, 39493-39504. [16] Saphire, A.C.S., Bobardt, M.D., Zhang, Z., David, G., Gallay, P.A.; Syndecans Serve as Attachment Receptors for Human Immunodeficiency Virus Type 1 on Macrophages. Journal of Virology, 2001, 75, 9187-9200. [17] De Clercq E.; Anti-HIV Drugs: 25 Compounds Approved within 25 Years after the Discovery of HIV. International Journal of Antimicrobial Agents, 2009, 33, 307–320. [18] Greene, W.C., Debyser, Z., Ikeda, Y., Freed, E.O., Stephens, E., Yonemoto, W., Buckheit, W.R., Esté, J.A., Cihlar, T.; Novel Targets for HIV Therapy. Antiviral Research, 2008, 80, 251-265. [19]Siegert, S., Schnierle, P., and Schnierle, B. S.; Novel Anti-Viral Therapy: Drugs that Block HIV Entry at Different Target Sites. Mini-Reviews in Medicinal Chemistry, 2006, 6, 557-562. [20] Johnson, M., Stallworth, T., Neilands, T.; The Drugs or the Disease? Causal Attributions of Symptoms Held by HIV-Positive Adults on HAART. AIDS and Behavior, 2003, 7, 109-117. [21] Lucas, G.M., Gallant, J.E., Moore, R.D.; Relationship between Drug Resistance and HIV-1 Disease Progression or Death in Patients Undergoing Resistance Testing. AIDS, 2004, 18, 1539-1548. [22] Veljkovic, V., Veljkovic, N., Glisic, S., Hob, M-W.; AIDS Vaccine: Efficacy, Safety and Ethics. Vaccine, 2008, 26, 3072-3077. [23] National Institute of Allergy and Infectious Diseases, National Institutes of Health, HIV/AIDS, http://www.niaid.nih.gov/topics/HIVAIDS/Understanding/Biology/Pages/ hidesImmuneSystem.aspx. Access date: 10/3/2011. Last updated: 6/2011. [24] Robb, M.L.; Failure of the Merck HIV Vaccine: An Uncertain Step Forward. The Lancet, 2008, 372, 1857-1858. [25] Smith, P.F., Ogundele, A., Forrest, A., Wilton, J., Salzwedel, K., Doto, J., Allaway, J.P., Martin, D.E.; Phase I and II Study of the Safety, Virologic Effect, and Pharmacokinetics/ Pharmacodynamics of Single-Dose 3-O-(3',3'-Dimethylsuccinyl) Betulinic Acid (Bevirimat) against Human Immunodeficiency Virus Infection. Antimicrobial Agents and Chemotherapy, 2007, 51, 3574-3581. [26] Cohen, John; The Emerging Race To Cure HIV Infection. Science, 2011, 332, 784789. [27] Lewin, S.R., Evans, V.A., Elliott, J.H., Spire, B., Chomont, N.; Finding a Cure for HIV: Will It Ever Be Achievable? Journal of the International AIDS Society, 2011, 14, 18. [28] Baba, M., Pauwels, R., Balzarini, J., Arnout, J., Desmyter, J., De Clercq, E.; Mechanism of Inhibitory Effect of Dextran Sulfate and Heparin on Replication of Human Immunodeficiency Virus in Vitro. Proceedings of the National Academy of Sciences of the United States of America, 1988, 85, 6132-6136. 142 [29] Baba, M., Snoeck, R., Pauwels, R., De Clercq, E.; Sulfated Polysaccharides Are Potent and Selective Inhibitors of Various Enveloped Viruses, Including Herpes Simplex Virus, Cytomegalovirus, Vesicular Stomatitis Virus, and Human Immunodeficiency Virus. Antimicrobial Agents and Chemotherapy, 1988, 32, 1742-1745. [30] Yang, D-W., Ohta, Y., Yamaguchi, S., Tsukada, Y., Haraguch, Y., Hoshino, H., Amagai, H., Kobayashi., I.; Sulfated Colominic Acid: An Antiviral Agent that Inhibits the Human Immunodeficiency Virus Type I in Vitro. Antiviral Research, 1996, 31, 95104. [31] Borman, S.; Multivalency: Strength in Numbers. Chemical and Engineering News, 2000, 78, 48-53. [32] Weis, W., Cusack, S., Paulson, J. C., Skehel, J. J.,Wiley, D. C.; Structure of the Influenza Virus Haemagglutinin Complexed with Its Receptor, Sialic Acid. Nature, 1988, 333, 426-431. [33] Landers, J.J., Cao, Z., Lee, I., Piehler, L.T., Myc, P.P., Myc, A., Hamouda, T., Andrzej T. Galecki, A.T., Baker, Jr., J.R.; Prevention of Influenza Pneumonitis by Sialic Acid-Conjugated Dendritic Polymers. The Journal of Infectious Diseases, 2002, 186, 1222-1230. [34] Aulenta, F., Hayes, W., Rannard, S.; Dendrimers: A New Class of Nanoscopic Containers and Delivery Devices. European Polymer Journal, 2003, 39, 1741-1771. [35] Dufes, C., Uchegbu, I.F., Schatzlein, A.G.; Dendrimers in Gene Delivery. Advanced Drug Delivery Reviews, 2005, 57, 2177-2202. [36] Tomalia, D. A.; Birth of a New Macromolecular Architecture: Dendrimers as Quantized Building Blocks for Nanoscale Synthetic Polymer Chemistry. Progress in Polymer Science, 2005, 30, 294-324. [37] Cloninger, M.J.; Biological Applications of Dendrimers. Current Opinion in Chemical Biology, 2002, 6, 742-748. [38] Zhang, X., Xu, H., Dong, Z., Wang, Y., Liu, J., Shen, J.; Highly Efficient Dendrimer-Based Mimic of Glutathione Peroxidase. Journal of the American Chemical Society, 2004, 126, 10556-10557. [39] Kensinger, R.D., Yowler, B.C., Benesi, A.J., Schengrund, C-L.; Synthesis of Novel, Multivalent Glycodendrimers as Ligands for HIV-1 gp120. Bioconjugate Chemistry, 2004, 15, 349-358. [40] Krishna, T. R., Narayanaswamy, R., Narayanaswamy, J.; Synthesis of Poly(propyl ether imine) Dendrimers and Evaluation of Their Cytotoxic Properties. Journal of Organic Chemistry, 2003, 68, 9694-9704. [41] Blackeye, R.; CSUS Unpublished results. [42] DeCampo, F., Lastecoueres, D., Vincent, J-M., Verlhac, J-B.; Copper(I) Complexes Mediated Cyclization Reaction of Unsaturated Ester under Fluoro Biphasic Procedure. Journal of Organic Chemistry, 1999, 64, 4969-4971. [43] Sarkar, R., Ahuja P., Vasos, P.R., Bornet , A., Wagnières, O., Bodenhausen, G.; Long-Lived Coherences for Line-Narrowing in High-Field NMR. Progress in Nuclear Magnetic Resonance Spectroscopy, 2011, 59, 83-90. [44] Chris Boesch, C.; Molecular Aspects of Magnetic Resonance Imaging 143 and Spectroscopy. Molecular Aspects of Medicine, 1999, 20, 185-318. [45] Macomber, R.S.; NMR Spectroscopy, Essential Theory and Practice. Harcourt Brace Jovanovich: pg. 12, 15, 1988. [46] Nudelman, A., Bechor, Y., Falb, E., Fischer, B., Wexler, B.A., Nudelman, A.; Acetyl Chloride-Methanol as a Convenient Reagent for: A) Quantitative Formation of Amine Hydrochlorides B) Carboxylate Ester Formation C) Mild Removal of N-t-BocProtective Group. Synthetic Communications, 1998, 28, 471-474. [47] Jam, F., Tullberg, M., Luthman, K. and Grøtli, M.; Microwave Assisted Synthesis of Spiro-2,5-Diketopiperazines. Tetrahedron, 2007, 63, 9881-9889. [48] Thygesen, M.B., Munch, H., Sauer, J., Cló, E., Jørgensen, M.R., Hindsgaul, O., Jensen, K.J.; Nucleophilic Catalysis of Carbohydrate Oxime Formation by Anilines. Journal of Organic Chemistry, 2010, 75, 1752-1755. [49] Baek, M-G. and Roy, R.; Synthesis and Protein Binding Properties of T-Antigen Containing GlycoPAMAM Dendrimers. Bioorganic & Medicinal Chemistry, 2002, 10, 11-17. [50] Cary, C.; Hardman, J., CSUS Unpublished work. [51] Isaacson, W.; Benjamin Franklin, An American Life. Simon & Schuster: pg. 104, 2003. [52] Newkome, G.R., Lin, X.; Symmetrical, Four-Directional, Poly(ether-amide) Cascade Polymers. Macromolecules, 1991, 24, 1443-1444. [53] Edupuganti, O.P., Renaudet, O., Defrancq, E., Dumy, P.; The Oxime Bond Formation as an Efficient Chemical Tool for the Preparation of 30, 50-Bifunctionalised Oligodeoxyribonucleotides. Bioorganic & Medicinal Chemistry Letters, 2004, 14, 28392842. [54] Rodriguez, R. A., Pan, P.-S., Pan, C.-M., Ravula, S., Lapera, S., Singh, E. K., Styers, T. J., Brown, J. D., Cajica, J., Parry, E., Otrubova, K., and McAlpine, S. R.; Synthesis of Second-Generation Sansalvamide A Derivatives: Novel Templates as Potential Antitumor Agents. Journal of Organic Chemistry, 2007, 72, 1980-2002. [55] Han, S.,Yoshida, T., Uryu, T.; Synthesis of a New Polylysine-Dendritic Oligosaccharide with Alkyl Spacer having Peptide Linkage. Carbohydrate Polymers, 2007, 69, 436-444. [56] Lonngren, J., Goldstein, I. J.; Carbohydrate antigens: Coupling Melibionic Acid to Bovine Serum Albumin using Water-Soluble Carbodiimide. Methods in Enzymology, 1978, 50, 160-162. [57] Caddick, S., Judd, D.B, Lewis, A.K. de. K., Reich, M.T., Williams, M.R.V.; A Generic Approach for the Catalytic Reduction of Nitriles. Tetrahedron, 2003, 59, 54175423.