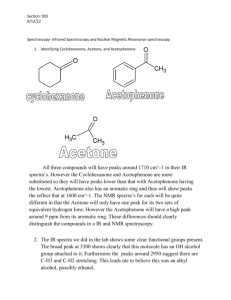
jws-pola.21931.tbl3
advertisement

SM Polymer Characterization Discussion Structural The one-dimensional proton NMR data for all co-PAAs, Zr pendent co-PAAs, and related compounds are collected in SM Table 1. Assignments and integrations are consistent with the proposed structures, with the following elaborations. Despite optimizing scan rate and time delay, polymer signals were less well resolved than for small molecules. This was due in part to the low solubility of precipitated powder samples in DMSO-d6, giving mediocre signal-to-noise ratios. Determination of coupling constants, and in some cases the coupling patterns themselves, was unreliable. However, the presence and mole percentage of MADA residues in the co-PAA backbone were indicated by additional amide proton signals that were much less intense than the amide proton signal from the other dianhydride residue. The MADA amide chemical shifts appeared slightly farther downfield due to the electron withdrawing effects of the additional carboxylic acid groups, and reflect the relative amounts of o-, m-, p- amide group isomerism. In addition, peaks matching those in the Zr(adsp)(dsp) spectrum confirm the presence of Zr pendent groups in all pendent polymers, and the integrations of non-overlapped pendent group peaks relative to polymer backbone peaks confirm the 10%(mol) pendent group concentration. Last, no proton signal from the amino groups of the free complex was observed. Coupled with the TLC results and observed color changes (see polymer synthesis, above), the DCC reaction product NMR spectra indicate that the appending reaction had occurred. Again, no signal appeared which could be assigned to a new amide group, which is expected to form during the appending reaction.1 We anticipate that strong electronic delocalization of the nitrogen’s electrons, 1 toward the carbonyl and toward the Zr,42 contribute to the amide proton’s lability. This lability could broaden the NMR signal, as for the carboxylic acid protons which likewise are not always observed. However, the change in chemical shifts of the unequal adsp imine protons following the DCC reaction, in conjunction with the dsp imine proton signal remaining at 8.7 ppm, is consistent with chemistry occurring at the amino group of the complex. Changes in the chemical shifts of the salicylidene protons meta- to the coordinated oxygen (5.8-5.6ppm), the proton signals that shift the most upon coordination,62 are also consistent with a change in the electron environment around the zirconium which would be expected upon changing the electron releasing amino group of Zr(adsp)(dsp) to an electron withdrawing amide group. Upon replacing the PMDA with ODPA in the co-PAA and Zr pendent co-PAA to give PAA 2 and ZrPAA 2, respectively, aromatic proton signals are found farther up field due to the electron releasing oxygen. Isomerism, arising from amide and carboxylic acid groups being meta and para to the bridging oxygen or visa versa, adds complexity to the amide proton region. Upon replacing 4,4’-ODA with 3,4’-ODA, certain aromatic peaks remain (7.7 and 7.0 ppm) while several new peaks appear due to the nonlinear metasubstituted ring. Upon replacing 3,4’-ODA with 1,3-APB, additional complexity is observed in the up field aromatic proton region (6.8-6.7 ppm) due to the presence of two electron releasing oxygen atoms and no electron withdrawing groups bonded to the central APB ring. Following thermal imidization of these polymers in the solid state, none were soluble in common organic solvents or water, so no NMR data for co-PIs were obtained. 2 Complementary to NMR, FT-IR spectra of co-PAAs were not very revealing due to the dominating presence of the solvent, NMP, which is also an amide; however, thermal imidization causes much of the solvent to be volatilized, allowing structural verification and extent of imidization to be determined for co-PIs. FT-IR data for all coPIs, Zr pendent co-PIs, and Zr(adsp)(dsp)63 are collected in SM Table 2. The spectral features common to all polyimides in this study include: the peaks in the 3600-3450 cm-1 range, which are attributed to water. The water arises from adsorption and/or is trapped during imidization. Similar peaks are observed in the homopolymer (without MADA) spectra. O-H stretching from carboxylic acid groups could absorb in this range, but the lack of peak intensity near 1650 cm-1 indicates that no amide carbonyl, hence no amic acid, is present after imidization. When MADA is present in the polymer backbone, a small, broad peak at 3350 cm-1 appears. [This peak becomes more intense as the mole percentage of MADA goes up, to be discussed more fully in the next paper in this series.] Assigning this peak as a carboxylic acid O-H stretch, rather than an amide N-H stretch, seems reasonable again since no amide carbonyl peak is apparent near 1650 cm-1. The peaks at ca. 1778, 1720, and 1379 cm-1 are consistent with imide ring formation.43 The peak(s) around 1250 cm-1 are consistent with the presence of at least one aromatic ringto-oxygen vibration. The aromatic ether structure is reputed to increase flexibility in an otherwise rigid, fused-ring PI.64,65 A striking feature in the IR spectra of all non-pendent co-PIs is the small peak at around 1860 cm-1. [This peak also increases in intensity with increasing mole percentage of MADA, as does the 3350 cm-1 peak.] Since the isoimide structure is not preferred at high temperature,61 this peak is assigned to the carbonyl vibration of an anhydride ring. 3 MADA, itself, has been shown to form a third anhydride ring at temperatures less than 300oC, the cure temperature used to prepare these PI films.1 The other peaks typically observed for the anhydride ring structure, around 1790 and 1220 cm-1 for asymmetric ring and C-O vibrations, respectively, are masked by peaks from the other (more prevalent) repeat units of the polymer backbone. The striking aspect of the 1860 cm-1 peak is that it is not observed in any of the Zr pendent co-PIs where the mole percentages of MADA and pendent groups are equal. The differences observed in the IR spectra of the co-PIs can be attributed to the different structures of the dianhydrides and diamines used in this study. Upon replacing PMDA in co-PI 1 with ODPA to give co-PI 2 (see SM Table 2), peaks due to the aromatic ring C-H vibrations appear at slightly lower frequency, and are more numerous due to the lower symmetry of the ODPA rings. The aromatic ring vibrations are likewise more numerous due to the lower symmetry. And, the aromatic ring-to-oxygen vibration peaks occur over a broader range since both 4,4’-ODA and ODPA moieties should display these. Upon replacing 4,4’-ODA in co-PI 2 with 3,4’-ODA to give co-PI 3, even more numerous aromatic ring vibrations and a broader aromatic ring-to-oxygen range are observed due to the still lower symmetry of the latter isomer. Upon replacing 3,4’-ODA in co-PI 3 with 1,3-APB to give co-PI 4, an aromatic peak at around 1505 cm-1 is no longer observed and the aromatic-to-oxygen range is a bit narrower, presumably due to the all meta (higher symmetry) bonding in the APB. Due to the low mole percentage of Zr pendent groups used in this study, the frequencies of all major peaks for Zr pendent co-PIs and non-pendent co-PIs match. Peaks due to imine C-H vibrations of the pendent groups are just barely perceptible for 4 ZrPI 3 and ZrPI 4, and certain other pendent group peaks are apparent when the structures of the co-PIs don’t mask them (see SM Table 2). Since the anhydride ring vibration is significantly weakened or absent from the ZrPI spectra (near 1860 cm-1, see above), pendent group attachment must interfere with ring formation. Thus, we conclude that pendent group attachment facilitated by DCC occurs at the MADA locations. The peak near 3350 cm-1 is observed in all ZrPI spectra [and increases with MADA and pendent group mole percentages, as for the co-PIs]. If the Zr pendent groups are attaching to only one carboxylic acid group of the MADA residue, then this peak can be assigned to the O-H vibration of the second carboxylic acid. 5