Taxonomy is the study of the classification of anything
advertisement

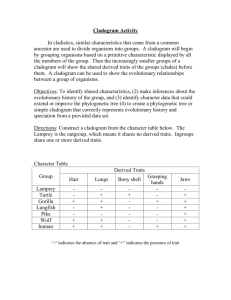
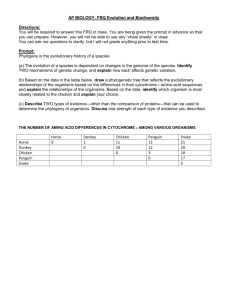
Taxonomy: Establishing Evolutionary Relationships The following lab contains information on classification and systematics. It will be the lab packet we are using for the week. You will NOT do a formal write-up for this lab. Rather, you will be completing the activities described in the lab. What to turn in: - A dichotomous key (one for each lab pair/group). - Pre-activity questions for making cladograms. (This will be checked.) - A cladogram of penguin species (one for each lab pair/group). - “Using computational analysis to determine evolutionary relationships.” One per person! You have a week after we do the cladogram lab to complete this! Taxonomy is the study of the classification of anything. In biology, taxonomy deals with the classification of organisms into groups based upon their similarities and differences. Biologists have generally classified organisms three Domains: Eukarya, Archaea, and Eubacteria. The Eukaryotes are divided into three Kingdoms, Animalia, Plantae, and Fungi. A fourth kingdrom, Protista, which included most single-celled eukaryotes, has been split into several sub-kingdoms, though these evolutionary relationships have yet to be resolved. Kingdoms are then further subdivided into large groups, or Phyla (Phylum, s.) consisting of those organisms that have very common, general characteristics. Phyla are subdivided into Classes, classes into Orders, orders into Families, families into Genera (Genus, s.), and genera into Species. Sometimes each of these subdivisions is further subdivided. In most cases, the subdivision is based on details of structure in the higher or larger categories and more on biochemical and genetic details in the lower categories. Scientific Classification has two main objectives: (1) to arrange or catalog organisms in a way which will make it easier for scientists to refer to them and, (2) to arrange organisms in a way that will demonstrate their evolutionary relationship to each other. (In the table, below, see example of a classification break-down using the human being.) Nomenclature is the science of naming newly identified, described, and classified organisms. Remember that Carolus Linnaeus developed the Binomial System of Nomenclature, or that system used to give an organism a two-word name using the genus and species from the taxonomic hierarchy. Species names are formatted properly as Genus species. The genus is capitalized, the species in lower case and the whole name is put in italics. QuickTime™ and a decompressor are needed to see this picture. Dichotomous Keys: The identification of biological organisms can be greatly simplified using tools such as dichotomous keys. A dichotomous key is an organized set of couplets of mutually exclusive characteristics of biological organisms. You simply compare the characteristics of an unknown organism against an appropriate dichotomous key. These keys will begin with general characteristics and lead to couplets indicating progressively specific characteristics. If the organism falls into one category, you go to the next indicated couplet. By following the key and making the correct choices, you should be able to identify your specimen to the indicated taxonomic level. Couplets can be organized in several forms. The couplets can be presented using numbers (numeric) or using letters (alphabetical). The couplets can be presented together (zoological style) or grouped by relationships (botanical style). Sample keys to some common beans used in the kitchen Numeric key with couplets presented together. The major advantage of this method of presentation is that both characteristics in a couple can be evaluated and compared very easily. a. Bean round Garbanzo bean 1b. Bean elliptical or oblong Go to 2 2a. Bean white White northern 2b. Bean has dark pigments Go to 3 3a. Bean evenly pigmented Go to 4 3b. Bean pigmentation mottled Pinto bean 4a. Bean black Black bean 4b. Bean reddish-brown Kidney bean Alphabetical key with couplets grouped by relationship. This key uses the same couplet choices as the key above. The choices within the first and succeeding couplets are separated to preserve the relationships between the characteristics. A. Bean elliptical or oblong Go to B B. Bean has dark pigments Go to C C. Bean color is solid Go to D C. Bean color is mottled: Pinto bean D. Bean is black: Black bean D. Bean is reddish-brown: Kidney bean B. Bean is white: White northern A. Bean is round: Garbanzo bean When you follow a dichotomous key, your task becomes simpler if you adhere to a few simple rules of thumb: Read both choices in a couplet carefully. Although the first description may seem to fit your sample, the second may apply even better. Keep notes telling what sequence of identification steps you followed. This will allow you to double-check your work later and indicate sources of mistakes, if they have been made. If you are unsure of which choice to make in a couplet, follow both forks (one at a time). After working through a couple of more couplets, it may become apparent that one fork does not fit your sample at all. Work with more than one sample if at all possible. This will allow you to tell whether the one you are looking at is typical or atypical. This is especially true when working with plants – examine more than one leaf, branch, cone, seed, flower, etc. When you have keyed out an organism, do not take your effort as the final result. Double check your identification scheme, using your notes. Find a type specimen (if available and usually in a museum somewhere) and compare your unknown to the type specimen. If a type specimen is unavailable, find a good description of the indicated taxonomic group and see if your unknown reflects this description. When reading a couplet, make sure you understand all of the terms used. The best keys will have a glossary of technical terms used in the key. If a glossary is unavailable, find a good reference work for the field (textbook, biological dictionary, etc.) to help you understand the term. When a measurement is indicated, make sure that you take the measurement using a calibrated scale. Do not “eyeball” it or take a guess. Exercise 1: Given a container of beans, use one of the dichotomous keys above to identify the beans. In your lab notebooks, write the common name of the beans with a brief description of each. Briefly discuss and write on the bottom of the card what steps you followed to arrive at your answer. Exercise 2: This exercise will help you to understand the process of classification of organisms by requiring you to construct your own classification scheme to a set of non- living objects. You will need a set of somewhat similar objects, such as a group of metal objects. As mentioned, above, describing an organism properly in understandable terms, is essential in the classification process. For each object in the entire group, write down a brief description of the object based on your observations of it. Use common descriptive terms (do not use specific terms, like “Phillips Head”, rather say “having an “x” or cross pattern on the top of the non pointed end.”) and measurements (if possible). It is not important that you give a name to each object at this time, but be sure to keep your descriptions and objects together. From the descriptions of all the objects, make a list of some of the common ways you have described them (e.g., size, shape, etc.). Give the number of objects that exhibit each characteristic. Constructing your own dichotomous key: This will be turned in, with one key for each group. Prepare a dichotomous key based on your descriptions. Each subdivision of your key should be represented by one couplet (pair, not three or four) of choices. These couplets should be close to opposites and should consider only one descriptor at a time, if at all possible. Names should be given to each object at this time if you have not done so already. Place the name at the end of the last descriptor that identifies that one item, only. Be creative (within reason) in giving each object a name. Remember, scientific names usually tell something about the object and are always written with the genus name capitalized and the species name in lower case. The species name is an adjective that describes the genus (e.g., Boltus longus) and they are italicized to set them apart from other words since they are usually in Latin or Greek. Systematic Approaches to Phylogeny Systematics is a discipline within biology whose goal is to determine the evolutionary history and relationships among organisms (termed phylogeny) and then the use of that phylogeny in classifying organisms. To achieve that goal, a systematist will utilize evidence from a wide variety of sources including paleontology, embryology, morphology, behavior, and molecular biology. Over the last few centuries, systematists have developed a number of different approaches for trying to show the relationships of organisms. As a result, different schemes have emerged for ordering these relationships into a classification scheme. * Cladistic (or phylogenetic) * Evolutionary (or synthetic) Cladistics (Phylogenetic Systematics) Cladistics is an approach to diagramming evolutionary relationships between taxa (taxon, s.), levels of organization, was developed. A clade is a branch of an evolutionary tree diagram. The basic assumption behind cladistics is that members of a group share a common evolutionary history, and are thus more "closely related" to one another than they are to other groups of organisms. Related groups of organisms are recognized because they share a set of unique features (apomorphies) which were not present in distant ancestors, but which are shared by most or all of the organisms within the group. Shared derived characteristics are called synapomorphies. (Shared ancestral characteristics are found in groups in that clade as well as groups outside that clade.) In cladistics, groupings do not depend on whether organisms share physical traits, but on their evolutionary relationships. In cladistic analyses, two organisms may share numerous characteristics but still be considered members of different groups. For example, a jellyfish, a sea star, and a human: jellyfish and sea stars both live in the ocean, have radial symmetry and are invertebrates, so phenetic analysis might place them together in a group. However, this would not reflect evolutionary relationships, as sea stars are actually more closely related to humans than they are to jellyfish, since both humans and sea stars have similar embryological development. Thus in cladistics, the emphasis is not upon the presence of all shared traits, but upon the presence of shared derived (apomorphic) traits. In the example above radial symmetry, aquatic habitat and invertebrate structure are all traits that are believed to have been present in the common ancestor of all animals, and so such traits are not considered to be very useful in determining relationships using cladistic analysis. Cladistic analyses have some pretty strict rules. For example, cladists always assume that new species arise by bifurcations of the original lineage (the lineage always splits in two), called branch points. Most cladists assume that the original ancestral species no longer exists after this bifurcation, so each branching event results in two new species. The two taxa that arise from the same branch point are called sister taxa. If more than two taxa arise from a branch point, it indicates scientists do not fully understand the ancestral relationships of all the species – also known as a polytomy. In addition, cladistic groupings must possess the following characteristics: 1. All species in a grouping must share a common ancestor. 2. All species derived from a common ancestor must be included in the taxon. The application of these requirements results in the following terms being used to describe the different ways in which groupings can be made: * A monophyletic grouping is one in which all species share a common ancestor and all species derived from that common ancestor are included. This is the only form of grouping accepted as valid by cladists. This is what is known as a rooted tree, since it contains the ancestral and descendent species of a common ancestor. (For example, turtles, lizards, crocodilians and birds are all derived from a shared common ancestor. Thus a monophyletic grouping would place all of these together, rather than placing birds into a separate group.) * A paraphyletic grouping is one in which all species share a common ancestor, but not all species derived from that common ancestor are included (for example, grouping turtles, lizards and crocodiles as "reptiles" and separating that grouping from the birds). * A polyphyletic grouping is one in which species that do not share an immediate common ancestor are lumped together, while excluding other members that would link them (for example, a hypothetical group the "lizmams" made by grouping together the lizards and the mammals). Thus, in cladistics, no matter how divergent in appearance B might be from C, relative to A, if B and C share a common ancestor that is not shared by A, then B and C must be grouped together and separated from lineage A. Steps for Constructing a Cladogram 1. Select a taxonomic group to be analyzed; for example, a group of vertebrates. 2. For each member of the group, determine some observable traits (characters), and note their "states" (a "character state" is one of two or more possible forms of that character). For example, for a character "fins," the possible states may be "present" and "absent." For the character "number of forelimb digits," possible states may be 1, 2, 3, 4, or 5. 3. For each character, determine which state is ancestral (plesiomorphic) and which is derived (apomorphic). This is usually done by comparison with a more distantly related organism termed the "outgroup." It is hypothesized that traits shared with the more distantly related organism(s) are likely to be "ancient" or plesiomorphic traits. Similarly, traits that differ from the outgroup are postulated to have arisen since the group being considered branched from its shared common ancestor with the outgroup, and thus are likely to be "derived" or apomorphic. 4. Group taxa by shared derived character states (synapomorphines). 5. When in doubt, choose the most parsimonious tree, trees that involve the fewest splits. (Parsimony = the simplest answer is probably the correct answer.) While similar structures may evolve independently in separate lineages facing similar selective pressures (convergent evolution), this is assumed to be a rare event. Most major structures (eyes, horns, tails, fur, etc.) are assumed to have evolved or to be lost only rarely. Thus, when in doubt, choose a pathway that minimizes the number of times a feature must be postulated to have arisen (or lost) separately. 6. In the event that two equally parsimonious trees can be produced, scientists use the principle of maximum likelihood, which states that the tree that shows roughly mutation rates in its branches is more likely than a tree showing vastly different mutation rates. In this example, frogs share all major traits with the outgroup (i.e., they show mostly ancestral or plesiomorphic traits), except that they have legs and slightly enlarged brains. These last two features are apomorphies that are widespread in vertebrates, animals with backbones and a dorsal nerve cord. Frogs are thus postulated to have branched from the main vertebrate lineage relatively early in the evolutionary process. Turtles show further modifications from the outgroup, most markedly the presence of a hard shelled egg, as well as an increased tendency toward larger brain size; therefore we would suggest that their lineage branched next from the ancestral lineage. The three remaining groups are characterized by an egg that develops inside the mother, suggesting that these three share a common ancestor not shared by frogs and turtles. Mice and kangaroos share similar hair amounts, while humans and kangaroos share a generally bipedal posture. So how do we know how to group these three organisms? Firstly, we would suspect that the possession of hair, even in reduced amounts, might link humans to kangaroos and mice. Secondly, we would look to the other traits possessed by these groups. Both mice and humans show placental development and thus lack a pouch. Thus we would tend to link these two groups more closely and the kangaroo more distantly. We would thus conclude that the cladogram for this group of organisms (minus the outgroup, which is not usually shown in these figures) should look something like the one below. You will note that, in this solution, the tendency toward a bipedal posture was postulated to have evolved twice; once in the marsupial lineage (kangaroos) and once in the placental lineage (humans). Such features are said to be analogous and to have resulted from convergent evolution. However, it is possible to draw the cladogram such that humans and kangaroos are postulated to have a common bipedal ancestor not shared by mice. However, in this solution, the tendency towards placental development, along with all the required anatomical changes (absence of a pouch etc.) must be postulated to have occurred twice (once in the mouse lineage and once in the human lineage). Such a solution would require more evolutionary steps than the sequence that we have proposed here and, consequently, would not be as efficient or "parsimonious" as our initial solution. Evolutionary (Synthetic) Systematics Like cladistics, evolutionary systematics is based strongly on evolutionary relationships. However, its supporters suggest that the degree of genetic differences between lineages should be used in addition to their genealogical (evolutionary) similarities when developing taxonomic classifications. The biggest difference between the two approaches is that, when creating groupings, evolutionary taxonomists seek to maximize the way in which the groupings they create communicate information about the group of organisms in that taxon (in the way the word "mammal" or "reptile" does for even an untrained person). This type of division is also intended to increase the ease with which a field biologist, or even someone coming in from outside, can retrieve information about a group of organisms of interest to him/her using the taxonomic divisions formed. Penguin Cladogram If you have seen the movies, “March of the Penguins”, “Happy Feet”, or “Surf’s Up!” you are probably familiar with at least a few species of penguins. (There are actually about 18 species, although there are a few additional subspecies.) All penguins are birds and they evolved from the same ancestral bird population as all other birds. Pre-activity Questions 1. What are some of the “bird-y” traits that the penguins share with all birds? These are the shared ancestral characteristics. 2. The 18 penguin species are all descended from the same ancestral penguin and share traits that they presumably inherited from the same “penguin-y” ancestor. What are some of the traits that penguin species seem to share? (Think beyond purely physical traits to their behavior and lifestyle as well.) These are shared derived characteristics for penguins. 3. Talk with your lab partners and try to think of the type of birds that are most closely related to the penguins. If you don’t actually know of a particular species or type of bird, perhaps you could predict its traits/behavior/lifestyle. This is identifying the outgroup. 4. What are some analogous adaptations penguins have with other species? Remember, analogous adaptations arise because of convergent evolution and do not show evidence of common ancestry. Making a Cladogram of the Penguin Species: Your group will receive an envelope with pictures of the 18 penguin species. Open it and spread the pictures out on your lab table. Some species will look similar to each other. We assume that these species have similar traits because they came from the same ancestral species. A cladogram shows which species share more common ancestry and which ones are more distantly related. A. Start by putting similar species into groups of species that seem closely related. You may want to look at the plumage and color patterns, body shape/size, beak shape, etc. These groups do not have to have the same number of species in them. Some species may not seem to fit well within a group – they may be left on their own for now. B. Take the largest group and try to break it into smaller groups until there are no more than two species in a group. You may begin arranging the species into the tree-like form of the cladogram. With four species, your cladogram could take one of the two forms below: 1 2 3 A 4 1 2 3 B 4 In arrangement A, species 1 and 2 seem more like each other than 3 and 4, and species 3 and 4 seem more like each other than they do 1 and 2. In arrangement B, 2, 3, and 4 seem more like each other and 1 seems to be the odd man out. Then, looking at 2, 3, and 4, species 3 and 4 seem more similar to each other than to species 2. If you had five species, the cladogram has more possible connections. Do your best to group them in a way that seems to make the most sense. C. Now that you have divided up your separate groups, you can start grouping the groups into larger groups. Again, there do not need to be equal numbers of groups or species in the larger groups. Just put those groups that seem to be more closely related next to each other. D. When you have all of the species grouped, you can draw the lines to make the whole cladogram. Please put all of the species at the same level on top of the cladogram and draw the connecting lines below. E. Compare your cladogram to other groups’ cladograms. If you and another group have differences, see if you can convince them that they are right. If you feel your cladogram is incorrect, you may rearrange it. Using Computational Analysis to Determine Evolutionary Relationships Instructions: A. Enter the NCBI home page at http://www.ncbi.nlm.nih.gov/. B. In the search window, enter “emperor penguin” and hit return. C. The next window gives the search results. The upper box shows written information about emperor penguins that is available from this site. The second box has information about genetic databases that contain data on emperor penguins. You will notice that there is not a lot there – not much work has been done with emperor penguins, it seems. D. We will look at core nucleotides – sequences for genes that show up in most organisms. These genes control basic cellular functions that you would expect all animals to possess. This database contains the sequences of only 13 core nucleotides in the emperor penguin. Click on the “Core Nucleotides” button. E. The eighth gene on this screen (its ID is DQ137225) is the gene sequence for a protein called cytochrome B (abbreviated “cytb”). Cytochrome proteins are involved in the release of energy from food in the mitochondria of the cell. One would expect this protein to be one that is shared by most, if not all animals. Click on the ID number. F. The next screen has information about this gene including who sequenced it and when it was sequenced. At the bottom of the screen, you actually get to see the 1008 base pairs (bp) that make up this gene. Scroll back up and click on the scientific name, Aptenodytes forsteri. G. This takes you to a page that is specifically about emperor penguins. The table on the right tells you what type of data are available through NCBI. At the bottom is a table with links out to other sources of information about the emperor penguin. In between, the full taxonomic lineage of the emperor penguin is listed. The family name of the penguins is Spheniscidae. Click on the family name. H. The next page lists the six penguin genera and the 18-20 species and subspecies. We will want to refer back to this listing, so either keep this window open or copy and paste the list into a Word document. The listing is in alphabetical order – it does not show the evolutionary relationships between the different genera and species. The NCBI site has tools that allow us to reconstruct a cladogram that shows the relatedness of these species based upon their genetic similarities. I. Go back to the NCBI home page and click the BLAST button right above the search box. BLAST stands for Basic Local Alignment Search Tool. BLAST is a computer program that can look at DNA from hundreds of species in the database at once and find which ones have the best match. At the top of the page are a dozen organisms for which we have complete genomes. (You may not recognize all of their scientific names, but nearly all are familiar organisms.) J. The program we will use is “nucleotide blast”. Click on the link and you will get the search page. In the “Enter Query Sequence” window where it says “Enter accession number...” enter the ID number “DQ137225”. In the “Choose Search Set”, click the “Others” button or else we will only compare the penguin to humans or to mice – we want to look at all available species. The pulldown menu below should default to “nucleotide collection (nr/nt)”. At the bottom, ask it to display the results in a new window and click the BLAST button. It will take a little while to perform the search for you. Be patient – it’s checking over 22.5 billion letters! K. On the new window, scroll past the gibberish at the top until you get to the table with the red bars. This table graphically represents the matches you got to the emperor penguin sequence. Below that, we have a table with descriptions of the sequences that matched in order of their similarity. Not surprisingly, the closest matches were with gene sequences from the same species Aptenodytes forsteri and from its close relative, the king penguin Aptenodytes patagonicus. As you scroll down the list, you may see the names of other penguin species that belong to different genera. However, there is a much better tool available to visualize the data. L. Under “other reports” (it’s in the box above the chart with the red lines), you will see “Distance Tree of Results”. Click it and maximize this new window to full size. Scroll down to the bottom and you should find the highlighted target sequence. What the program has done is to construct a cladogram of the results. The distance tree is arranged like a cladogram in that it groups protein sequences on the same branches if they share a great deal of similarity. If the emperor and king penguins evolved from the same ancestral population of penguins, we would expect their DNA to be more like each other’s than any other species. Looking at this one cytochrome gene, that appears to be the case. Not only are these two species similar in plumage, physical structure, behavior, calls, diet, and habitat, their cytochrome b gene sequence is similar as well. All of these lines of evidence support the fact that these species evolved from the same common ancestor. Analysis: 1. Use the BLAST tree to construct a cladogram of the penguin species that are listed. (If you find two or more “twigs” on a branch that share the same species name, just lump them together as one twig.) 2. What are some of the traits shared by penguins that they all must have inherited from their common ancestor? (Think beyond just physical traits, too.) 3. If you were to expand your cladogram, it would have to start including birds that are NOT penguins. What modern bird(s) do you suppose are most closely related to penguins? Go back to your BLAST tree and find the branch that connects closest to the penguin branch. You may need to use Google or some other search engine to find out what these scientific names mean. What birds seem to be the most closely related to penguins? 4. Pick a different animal and use a cytochrome b or c BLAST sequence to construct a cladogram of its most closely related genera and species. Try to stick to complete sequences instead of partial sequences, if possible. Include at least six separate species and/or subspecies in your cladogram.