Supplemental Material
advertisement

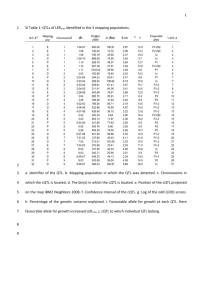
Makarevitch et al., Supplemental on-line text MATERIALS AND METHODS Plant materials and tissue collection: The zmet2-m1 allele was generated by Mu transposon mutagenesis and contains a Mu insertion in exon 18 that results in a loss-offunction mutation (Papa et al. 2001). This allele was backcrossed into the Mo17, B73, and W22 r-g inbred lines for six generations and then self-pollinated for two generations to produce homozygous near-isogenic lines of Mo17, B73 and W22 r-g that are homozygous for the zmet2-m1 allele. Three biological replicates of the B73 and B73 zmet2-m1 genotypes were grown sequentially over a 5 week period during May and June 2005. Three additional biological replicates involving the B73, B73 zmet2-m1, Mo17 and Mo17 zmet2-m1 genotypes were grown during January and February 2006. The plants were grown using standard greenhouse conditions (1:1 mix of autocalaved field soil and MetroMix; 16 hours light and 8 hours dark; daytime temperature of 30 0C and night temperature of 22 0C) and were sampled for gene expression on the 11th day after planting between 8 am and 9 am. The plants were cut immediately above the highest root, thus all above-ground tissues and meristems were collected. For each biological replicate, the seeds were planted such that one seed of each genotype was present in every pot. Eight pots were selected for tissue collection resulting in sampling of eight plants per genotype in each replicate. The sampled tissues were flash frozen in liquid nitrogen and stored at -80C prior to RNA isolation. For the analysis of plants that were segregating for the zmet2-m1 allele seeds derived from a self-pollination of the backcross six generation were planted and genotyped. Genotyping was performed using the primers Zmet2F23, ZmetR23 and 9242 (see Table SOM1 for all primer sequences). A single band of ~500 bp is amplified in homozygous wild-type plants while two bands of ~400 and ~200bp are amplified from homozygous zmet2-m1 plants. All three bands are observed in heterozygous individuals. RNA isolation and microarray hybridization: RNA isolation and Affymetrix (Santa Clara, CA, USA) microarray hybridizations were performed as described (STUPAR and SPRINGER 2006) with several modifications. Briefly, tissues from 8 seedlings per genotype per biological replicate were pooled and ground in liquid nitrogen. RNAs were extracted using Trizol reagent according to the manufacturer’s instructions (Invitrogen Corp., Carlsbad CA) and purified using the RNeasy kit, according to the manufactures instructions (Qiagen Corp., Valencia, CA). The quality and quantity of all purified RNA samples were assessed using agarose gel electrophoresis and the Nanodrop spectrophotometer (Nanodrop Technologies, Montchanin, DE). Affymetrix microarray hybridizations were performed for three biological replicates for each of four genotypes: mutants B73 zmet2-m1 and Mo17 zmet2-m1, and inbreds Mo17 and B73 and for three additional replicates for B73 and B73 zmet2-m1. Eight g of total RNA was labeled for each hybridization using the One-Cycle cDNA Synthesis Kit, according to the manufacturer’s instructions (Affymetrix, Santa Clara CA) and sent to the University of Minnesota Microarray Facility for hybridization to the Maize GeneChip array produced by Affymetrix. Spotted long oligonucleotide array hybridizations were performed for three biological replicates for the mutants B73 zmet2-m1 and inbred B73 genotypes using a subset of the RNA samples used for Affymetrix hybridizations. For this experiment, dual channel (Cy3 and Cy5 per slide) studies were performed with complete dye swapping. Total RNA from each biological replicate was indirectly labeled using Ambion’s Aminoallyl Message Amp II kit (Ambion, Austin TX). This procedure requires 1.5 µg of total RNA to incubate at 70°C in a thermal cycler for 10 min with 1 µl of T7 Oligo (dT) Primer and 6 µl of nuclease-free water. Then, a first strand cDNA reaction was carried out with by adding 1 µl of 10x first strand buffer, 0.5 µl of Ribo. Inhibitor, 2 µl of dNTP, 0.5 µl of Array Script to the total RNA T7 oligo (dt) primer mix. This was followed by 2 hours incubation in a thermal cycler at 42°C. A second strand cDNA reaction was then performed by adding 31.5 µl of nuclease free H2O, 5 µl 10x 2nd strand buffer, 2 µl dNTP 1 µl DNA polymerase and 0.5 µl RNase H to the 1st strand cDNA samples. The samples were incubated in a thermal cycler at 16°C for 2 hours. The cDNA was purified using the cDNA purification kit supplied with the Message Amp-II kit and the procedure used here were the same as describe in this product’s manual. Clean cDNA was checked by Nanodrop spectrophotometer (Nanodrop Technologies, Montchanin, DE) to ensure proper concentration (5-10 ng/ µl). Cleaned cDNA was used for an in vitro transcription reaction to produce aminoallyl labeled cRNA. The in vitro transcription reaction required 1.5 µl of aaUTP (50 mM), 6 µl of ATP, CTP, and GTP Mix, 0.5 µl of UTP (75 mM), 2 µl of T7 10x Rxn buffer, 2µl of T7 enzyme for 8 µl of each cleaned cDNA sample. Each sample was incubated in 0.2 ml 8-tube strips from Molecular BioProducs (catalog # 3418) for 10 hours at 37°C in a Forma Orbital shaker (Thermo Electron Corporation). The reaction was then stopped by adding 80 µL nuclease-free water to each cRNA sample bringing the final volume to 100 µL. The cRNA was purified using the cRNA purification kit supplied with the Message Amp-II kit following the protocol provided in the manual. The concentration of cRNA in each sample was then measured using a Nanodrop spectrophotometer (Nanodrop Technologies, Montchanin, DE), and 6 µg from each sample were dried using a CentriVap benchtop centrifugal concentrator (Labconco Corporation, MO). The dried samples were dissolved in 5 µL of NaHCO3 (200mM Na2CO3, 200mM NaHCO3 and pH 9.0) allowed to rest at room temperature for 20 minutes and mixed by flicking several times. Thereafter, enough Cy3 and Cy5 monoreactive dye (Amersham Pharmacia; Cat# PA23001 and PA25001 respectively) were added react with 7.5 nmol amino groups. In the case of these dyes 22 µl of dimethyl sulfoxide (DMSO) were added to each vial provided by Amersham and distributed to 4 of the cRNA samples by adding 5 dye-DMSO. The coupling reactions were allowed to incubate in complete dark for 2 hours at room temperature. Excess dye was quenched by using 4.5 µL of 4M hydroxylamine for 15 minutes at room temperature. The removal of unincorporated dye was accomplished using the Qiagen RNAeasy Mini elute columns and buffers (Qiagen Cat # 74204) following the manual procedure. The preparation of the long oligonucleotide arrays for hybridization has been previously reported. The labeled targets were measured for concentration of RNA and dye incorporation with Nanodrop spectrophotometer (Nanodrop Technologies, Montchanin, DE). Each array was hybridized with 2 µg of cRNA from one genotype and 2 µg from the other labeled with different Cy dyes. The hybridization solutions per slide contained 4 µg of cRNA, 8 µl of 20X SSC, 2.4 µl of Liquid Block (Amersham; Cat # RPN3601) 3.2 µl of 2% SDS and nuclease free H2O to 75 µl. The hybridization mixtures containing the labeled cRNA targets were denatured at 65°C for 5 minutes and then placed on ice immediately. The slides were covered with lifterSlip cover slips (Catalog #: 24X601-2- 4733) and the hybridization mixture was introduced by capillary action and evenly distributed throughout the slide. The spotted long oligonucleotide arrays were hybridized in Corning hybridization chambers (Corning life science, NY) at 55°C for 10 hours, in dark conditions, in a Forma Orbital shaker (Thermo Electron Corporation). Subsequently, the slides were washed by immersion and slow shaking in 2x SSC, 0.1% SDS at 55°C for 5 minutes followed by 5 minutes washing in 0.5x SSC and 5 minutes in 0.08% SDS, Finally, the slides were dipped 10 times in 0.05x SSC and spun at 1000 rpm for 4 minutes to dry. The dried arrays were immediately scanned in an Axon scanner GenePix 4000B (Molecular Devices Corporation) as recommended by this product’s manual. RESULTS Analysis of differential expression in B73 and B73 zmet2-m1 using long oligo spotted array platform: Due to the lack of repetitive elements and transposons on the Affymetrix platform and to test a larger set of genes for differential transcript accumulation, we hybridized the same RNA samples from three replicates of B73 and B73 zmet2-m1 to 70-mer long oligonucleotide microarrays (www.maizearray.org). Of the 56,309 probe sets present on the array, 179 probe sets were found to be differentially expressed between B73 wild-type and B73 zmet2-m1 mutant plants with 81 being upregulated and 98 down-regulated in B73 zmet2-m1 plants (Figure SOM2, Table SOM3). This represents a small proportion of the probe sets (0.3%), similar to the proportion identified by the Affymetrix analysis. Sixty-two out of 80 genes identified as differentially expressed in B73 versus B73 zmet2-m1 comparison by Affymetrix platform were also present on the spotted long oligonucleotide array. Nineteen of these 62 genes (31%) were also identified as differentially expressed using the long oligonucleotide microarray platform. Of the remaining 43 genes, 30 were altered in the same direction on both microarray platforms but did not pass the strict statistical criteria in the analysis of long oligonucleotide array data (Table SOM2). The normalized signals from both platforms showed good correlation for both genotypes tested suggesting that data received from both platforms were in agreement (Figure SOM3).