Population samples: We sampled populations from across much of
advertisement

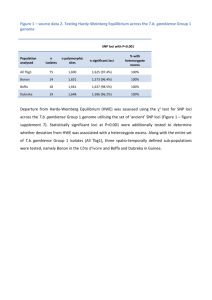
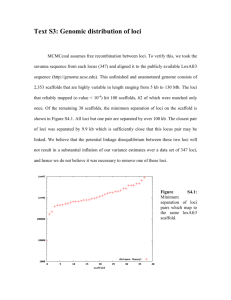
Supporting Information Methods S1 Population samples We sampled populations throughout much of the range of Eichhornia paniculata, including N.E. Brazil, Jamaica and Cuba in 2005, 2007 and 2008, respectively (Supporting Information Table S1). Mature fruit were obtained from up to 20 openpollinated maternal families in each population. We also estimated the frequencies of the three floral morphs and the size of populations. Material from both Mexico and Nicaragua was collected as a bulk sample from an unknown number of families and therefore these two populations were not included in our analyses of population differentiation. We germinated seed and grew plants for DNA extraction under glasshouse conditions at Toronto following standard protocols. Marker development We developed nuclear DNA markers based on EST sequences collected from a cDNA library from leaf tissue from single plants. We purified poly-adenylated RNA from total RNA using the Ambion Micro Poly(A) Purist kit and reverse transcribed the mRNA using the InVitrogen Superscript cDNA synthesis kit. We cloned cDNA into E. coli and sequenced approx. 480 clones. From these clones, we selected sequences that aligned well to known nuclear sequences from other plants and designed primers to amplify both coding regions and intron sequence when possible. We initially tested the primers by sequencing the loci in highly inbred individuals with the rationale that there should be few heterozygous sites. Loci with heterozygous sites were excluded as likely paralogs. From these screens we chose 10 EST-derived nuclear markers of the following lengths; EP0001- 530bp, EP0141 - 825bp, EP0143 - 846bp, EP0144 - 822bp, EP0188 - 383bp, EP0222 - 481bp, EP0267 - 561, EP0285 - 827bp, EP0314 - 752bp and EP0317 - 651bp. Amplification and sequencing We extracted DNA from each of 229 individuals which was used to PCR amplify the 10 loci in each individual. We sequenced both forward and reverse strands with an ABI 3730XL fluorescent-based capillary sequencer at the Centre for Applied Genomics facility at Sick Kids Hospital, Toronto, Ontario, Canada. We assembled and aligned sequences using Sequencher 4.7 and edited chromatographs and alignments manually to ensure that all base calls and polymorphisms, including heterozygotes, were reliably scored Sequence analysis We generated neighbour networks using the program SplitsTree. We estimated distance among individuals as an uncorrected number of differences and calculated the network using the NeighborNet method. Support for the network was estimated with 10,000 bootstrap replicates and nodes with greater than 70% support are indicated on the network (Fig. 3). We displayed the resulting network using one individual per population that was randomly sampled using a custom computer script. In addition, we generated the network for all 229 individuals to ensure that the network illustrated for single individuals per population (Fig. 3) was representative of patterns across all individuals. Using the program SITES we calculated pairwise FST for each of the 10 loci across all populations and groups (Caribbean, Brazilian trimorphic outcrossing, Brazilian monomorphic selfing). We calculated mean FST by averaging pairwise FST estimates across all loci. For each pairing the program SITES also provided the number of polymorphism within each population or group, the number of polymorphisms segregating in both, and the number of fixed differences between them.