Experiment 6 - Computational study of carbocation stability and SN1
advertisement

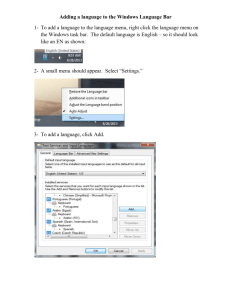
CHEM 350 Principles of Organic Chemistry I Lab Prof. T. Nalli, WSU Experiment 6 - Computational study of carbocation stability and SN1 reaction rates. References (1) Smith, Chapter 7. 14. (2) Pavia et. al., Expt 19B&D and pp 160-168. (3) Screttas, C. G. J. Org. Chem. 1980, 45, 333-326. OVERVIEW We will carry out experiments 19B and 19D in Pavia using Hyperchem molecular modeling software. The molecular orbital calculations being used make fewer assumptions and provide more reliable molecular energies that the molecular mechanics method used in previous modeling labs. You will need to install Hyperchem on your computer: (You must be logged on to the campus network to install and run Hyperchem. 1. Go to \\appsrv1\apps\hyper75install\install\Hyper75 and run the setup file. When prompted enter your user name, “wsu” as your organization, and “edutechnology” as the dealer. The serial number must be entered as 12-750-1601800013. Choose “networked installation” and “software license key”. 2. Go up two folders (to \\Appsrv1\apps\hyper75install\install and run the “HyperChem752Update” file followed by the “LSHOST check” file. 3. Go to \\Appsrv1\apps\Hyper75\Program and click and drag the CHEM (a green beaker) on to your desktop. Double click on your new HyperChem shortcut and the program should start. (You may be asked to once again enter the serial number, etc. Part 1 – (Expt 19B, Part 1) Gas Phase SN1 (No Solvent) General Instructions. Construct the compound of interest by using the “draw” tool (left most icon in the toolbar at top) to click and drag C-C bonds just as you would in Chem3D (one click gives you a single carbon). Attach Br by first selecting Br as the “default element” under the “build” menu. (You will need to change this back to carbon before building other molecules.) Then click, drag, and release from the carbon to which the Br needs to be bonded. Finally, select “Add H and Model Build” under the “Build” menu. (There is no “undo” capability in Hyperchem, but if you make a mistake you can delete the offending atoms by right clicking on them with the draw tool.) As directed in Pavia, we will be doing semi-empirical MO calculations using the AM1 parameter set. Set up Hyperchem to do this by selecting “Semi-empirical” under the “Setup” menu and then “AM1”. To compute the energy of each molecule once built, select “Geometry Optimization” under the “Compute” menu and then “OK”. (An RMS gradient of 0.01 and the PolakRibiere energy minimization algorithm should both work fine for our purposes.) The energy displayed by the program once the calculation is completed represents the Standard Enthalpy of Formation of the molecule in kcal/mol. Suggested Procedures 1. Calculate the energy of each of the four alkyl bromides in turn. Save each molecule once the calculation is complete (you will need them later) and then create the next one by replacing one H with a CH3. 2. Now calculate the energy of the carbocations. Pull up each saved RBr file in turn and first remove the bromine atom (right click on it). Under “Build”, choose “Explicit Hydrogens” and “Allow Arbitrary Valence”. Then go to the “Setup” menu and select “Semi-empirical”, “Options”. This is where you set the charge to +1. Also set the spin multiplicity to 1. Calculate the energy of the carbocation using “Geometry Optimization” as before. (Also note your observations on the geometry of the optimized carbocation.) (Also save each carbocation file before going on to the next calculation!) 3. Lastly, compute the energy of a bromide ion. Set the charge to –1 in the same manner as in step 2. Part 2 – (Expt 19B, Part 2) Solution Phase SN1 (H2O Solvent) For this part, we will need to repeat all of the calculations of part 1 but this time including a surrounding matrix of water molecules. In Hyperchem, this is accomplished by placing the molecule in a defined box. First reopen the file for the structure to be calculated. Then under “Setup”, choose “Periodic Box”. Set the periodic box dimension for all directions to nine angstroms. Now, as before compute the energy using “Geometry Optimization”. Set the maximum number of cycles to 250. The calculations will take some time- be patient. Part 3 – (Expt 19D, Part 1) Carbocation Electrostatic Potential Maps Unfortunately, Hyperchem 7.5 no longer allows the generation of electrostatic potential maps that can be compared meaningfully from molecule to molecule. Specifically it will not allow the setting of color values (as described in Pavia) to the same range for the four carbocations to be examined. (Hyperchem now automatically determines the range of color values to be used for each molecule). Suffice it to say the maps would come out exactly as pictured on p 249, Fig 7.17 in Smith. No matter, what we will do is better…obtain quantitative predictions of the charge on each carbon Reopen each carbocation structure from part 1. Under the “Display” menu select “Labels”. Under “Atoms” select “charge”. Record the charge of each carbon atom as data in your notebook. Pay special attention to the charge on the carbocation carbon, i.e, the carbon with formal charge = +1. Part 4 – (Expt 19D, Part 2) Allyl Cation Electrostatic Potential Map Build and optimize the allyl cation, CH2=CH-CH2+, using the techniques learned earlier in the lab. Note your observations on the optimized structure and then display and record the atomic charges using the same procedures as in part 3 above.