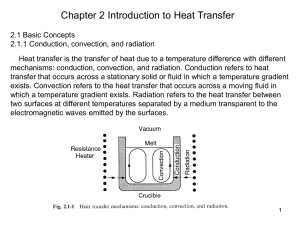
Mj_MIAME_information
advertisement

Experimental design These experiments were designed to study the effect of heat shock and cold shock on expression profiles of a hyperthermophilic archaeon after a twenty-minute temperature shock. When cells entered the middle of the exponential growth phase at the optimal growth temperature of 85C, they were transferred from an 85C water bath to a 95C water bath or a 65C water bath. Two biological replicates were obtained under both heat shock and cold shock conditions. For each biological replicate, we performed a total of six technical replicates consisting of three pairs of flip-dye experiments. As for the reference sample, a pooled population of cDNA from three biological replicates consisting of cells growing in the mid-exponential phase at 85C was used. All hybridizations were performed against this reference. Samples used, extract preparation and labeling Methanococcus jannaschii was obtained from the Oregon Collection of Methanogens (David Boone). Cells were cultivated in 125ml clear serum bottles (Wheaton) containing 30 ml of media as described previously (Miller et al., Appl Environ Microbiol 1988) with Na2S as the reducing agent. To inoculate the media, 3ml of culture stored anaerobically at room temperature was added, and the bottle was pressurized to 30 psi with a 4:1 v/v mixture of H2 and CO2 substrate. Bottles were placed in a reciprocal shaking water bath (Precision Scientific Model 25) at 200 oscillations per min and 85C. Growth was followed by measurement of optical density at 660 nm (Tsao et al., Biotechnol Bioeng 1994). In the mid-exponential phase, serum bottles were transferred from the 85C water bath to another water bath agitating at 200 oscillations per min and 65C or 95C. After twenty minutes, 20ml of cells were harvested and RNA extracted. Cells were passed through a 0.45 m nitrocellulose filter (Millipore) for collection. The filter was placed into cold phenol with 1% SDS and 0.1M Na-acetate and bead-beat (BioSpec Products, Inc.) for 5 minutes at full speed. The aqueous layer was removed and re-extracted with phenol, and the sample was then precipitated overnight at -20C with 3M sodium acetate and isopropanol. Precipitated RNA was re-suspended and DNasetreated (Invitrogen). The RNA was purified with the RNeasy kit (Qiagen), and RNA quality was assessed using the BioAnalyzer 2100 (Agilent). RNA was amino-allyl labeled with NHS-Cy dyes. 2 g of RNA and 2 l of random hexamers (Invitrogen) were combined, incubated at 70C for 10 minutes, and snap freezed in a dry-ice/ethanol bath for 30 sec. cDNA synthesis was carried out at 42C overnight with SuperScript II (Invitrogen), 0.01M DTT, and 0.012 aa-dNTP mix (12.5 mM dATP/dCTP/dGTP (Invitrogen), 4.16 mM dTTP (Invitrogen), and 8.33 mM aadUTP (Ambion)). RNA template was hydrolyzed with 10 l 1M NaOH and 10 l 0.5M EDTA followed by incubation at 65C for 15 min. cDNA was separated from reaction reagents using a Microcon 30 spin column (Millipore) and lyophilized to dryness using a Speed Vac (Savant). For NHS-Cy dye coupling, samples were re-suspended in 4.5 l 0.1M sodium carbonate pH 9.0 and 4.5 l of either NHS-Cy3 or NHS-Cy5 (Amersham Pharmacia) and allowed to couple to the cDNA for one hour at room temperature in the dark. The coupling reaction was quenched with 4.5 l hydroxylamine and incubated in the dark for 15 minutes at room temperature. 35 l of 100 mM sodium acetate pH 5.2 was added ,and unincorporated dye was removed using the QIAquick PCR purification kit (Qiagen). Coupled targets were analyzed using a spectrophotometer (Beckman) to determine how much dye is incorporated and how many nucleotides there is per incorporated dye. Targets with dye incorporation over 150 pmol and nucleotides per dye ratios below 50 were used for subsequent hybridization. These Cy3 and Cy5-labeled cDNA were lyophilized to dryness in a Speed Vac (Savant). Hybridization procedures and parameters Slides were pre-hybridized in filter-sterilized buffer (5 SSC, 0.1% SDS, 10 mg/ml BSA) at 42C for at least one hour prior to use. Slides were then washed in MilliQ H2O for ten minutes, in isopropanol for six minutes, and spun dry. Lyophilized, labeled cDNA targets were re-suspended in 30 l of hybridization buffer (50% formamide, 5 SSC, 0.1% SDS, 0.2 mg/ml BSA) for 15 min at room temperature, and Cy5 and Cy3-labeled targets were combined. Targets were then heated for 10 min at 95C and cooled on ice for 30 sec. A clean 25mm 60 mm glass lifterslip (Erie Scientific) was placed onto each pre-hybridized slide, and target was applied using capillary action. Slides were placed in hybridization chambers (Corning) and allowed to hybridize at 42C overnight. Post-hybridization washes were performed in an aluminum-foil wrapped glass dish (VWR) on an orbital shaker. The first wash was performed in 1 SSC, 0.1% SDS at 42C for 5 min. The second wash was performed in 0.1 SSC, 0.1% SDS for 5 min and the final three washes were performed in 0.1 SSC for 5 min at room temperature. Measurement data and specifications Slides were scanned on a GenePix 4000 dual-color confocal laser scanner (Axon) with PMT values set between 700 and 850. Paired 16 bit tiff files at 532 nm and 635 nm were saved for each slide. Spots were identified using SpotFinder (v2.2.2, TIGR) with an output of local background-subtracted Cy3 and Cy5-values. Data for spots with more than fifty non-saturated pixels, a spot area/spot perimeter ratio of greater than r/2 (r=radius of spot), and 50% of pixel spot intensities greater than 2*median(background) were considered good and kept for analysis. Another criterion for data usability was PCR scores. PCR products were run on gels and scored into several categories: failed=0, strong=1, weak=4, smear=5 or misprime=6 (see “Mj cDNA array description file.xls” for this information). Spots containing PCR products scored as strong or weak and passing the SpotFinder criteria were normalized. Normalization and transformation was performed using MIDAS (v2.16, TIGR). Data was print-tip lowess normalized with a smoothing parameter of 0.33 (the percent of data used to calculate the lowess factor) and then standard deviation regularized across printtip groups (so that each group has the same spread). Pairs of flip-dye data (here called file 1 and file 2 as an example) were then subjected to a flip-dye consistency check in which log2((R1*R2)/(G1*G2)) values are calculated (where R1/R2=Cy5-intensity values in file 1 and 2, respectively, and G1/G2=Cy3-intensity values in file 1 and 2, respectively) and data outside of two standard deviations are zeroed out. Finally, hybridization data within either the heat shock or cold shock experiments were standard deviation regularized across slides. The output files contain standard deviation regularized Cy3 and Cy5-values that are the geometric means of values found in each pair of flip-dye files (Cy3=G1*R2 and Cy5=R1*G2). Within both the heat and cold shock experiments, we are now left with six files (two biological replicates with three pairs of dye-swap experiments that have been combined in one data file). These data were inputted into MEV (v2.2, TIGR) for significance analysis. T-values were calculated for spots with data in at least four of the six data files. Expression ratios meeting the unadjusted p-value cutoff for the normal t-distribution of 0.01 were outputted. For data in the paper, in-slide replicate data for genes meeting the pvalue cutoff of 0.01 were arithmetically averaged together to report a fold change for each gene. Genes with fold-changes of greater than or equal to two were considered differentially-expressed. The smallest and largest p-value out of all the in-slide replicate data is also reported for each gene. Data meeting these cutoffs is contained in the file “Genes_meeting_2-fold_and_p_less_than_0.01_cutoff.xls.” See Table 1 for the list of other outputted data files. Array Design cDNA spotted glass arrays were printed at the Institute for Genomic Research (TIGR) as follows. Primers (Invitrogen) were designed for all 1,738 ORFs. PCR reactions were performed with 1 ng of genomic DNA and Platinum Taq polymerase (Invitrogen). Products were detected on agarose gel and cleaned and concentrated using 96-well PCR filter plates (Millipore). Resuspended products were again detected on agarose gel and scored into several categories: failed, strong, lost-on-filter, weak, smear, or misprime. Products scored as “lost-on-filter” were reamplified and precipitated using EtOH and 3M sodium acetate at -80C. Only data for spots scored as strong or weak were used in the array analysis (about 99% of the ORFs). PCR products were arrayed into 384-well plates with 50% DMSO (Sigma). Ultra Gaps slides (Corning) were then printed with these products using a MD-3 robot (Amersham Pharmacia) with print-tips arrayed in a 2 by 12 format. Eight copies of genes participating in methanogenesis and four copies of the rest of the genome were printed. Printed slides were placed in metal slide racks (VWR) and baked at 80C for two hours. Table 1. Name of files outputted in each step of the array analysis. 85E = 85C midexponential sample. 65-1 and 65-2 = two biological replicates for the 65C cold shock (65TS). 95-1 and 95-2 = two biological replicates for the 95C heat shock (95TS). There are six technical replicates for each biological replicate consisting of three sets of flip-dye pairs. Tav files are arranged as follows: 1st six columns are the slide coordinates, 7th and 8th columns are the Cy3 and Cy5 values, respectively, 9th and 10th columns are the Spotfinder flags (see Spotfinder documentation, TIGR), 11th column is the ORF, 12th column is the PCR score, and the 13th column is the common name. In the Spotfinder outputs, 1st sample in the file name corresponds to the Cy3 value and 2nd sample in the file name corresponds to the Cy5 value. In the MIDAS outputs, the 7th column consists of the temperature shock sample and the 8th column consists of the 85C exponential sample. Spotfinder MIDAS MEV MJ_7884(85E_vs_65-1).tav 1st_set_MJ_7884(85E_vs_651)_MJ_7881(65-1_vs_85E).tav MJ_7881(65-1_vs_85E).tav MJ_3987(85E_vs_65-1).tav 2nd_set_MJ_3987(85E_vs_651)_MJ_3985(65-1_vs_85E).tav MJ_3985(65-1_vs_85E).tav MJ_7939(85E_vs_65-1).tav 3rd_set_MJ_7939(85E_vs_651)_MJ_7938(65-1_vs_85E).tav MJ_7938(65-1_vs_85E).tav 65C_significant_genes.xls MJ_7885(85E_vs_65-2).tav 1st_set_MJ_7885(85E_vs_652)_MJ_3611(65-2_vs_85E).tav MJ_3611(65-2_vs_85E).tav MJ_3941(85E_vs_65-2).tav 2nd_set_MJ_3941(85E_vs_652)_MJ_3942(65-2_vs_85E).tav MJ_3942(65-2_vs_85E).tav MJ_7941(85E_vs_65-2).tav 3rd_set_MJ_7941(85E_vs_652)_MJ_7940(65-2_vs_85E).tav MJ_7940(65-2_vs_85E).tav MJ_3608(85E_vs_95-1).tav 1st_set_MJ_3608(85E_vs_951)_MJ_3609(95-1_vs_85E).tav MJ_3609(95-1_vs_85E).tav MJ_3986(85E_vs_95-1).tav 2nd_set_MJ_3986(85E_vs_951)_MJ_3984(95-1_vs_85E).tav MJ_3984(95-1_vs_85E).tav MJ_7777(85E_vs_95-1).tav 3rd_set_MJ_7777(85E_vs_951)_MJ_7776(95-1_vs_85E).tav MJ_7776(95-1_vs_85E).tav 95C_significant_genes.xls MJ_3612(85E_vs_95-2).tav 1st_set_MJ_3612(85E_vs_952)_MJ_3610(95-2_vs_85E).tav MJ_3610(95-2_vs_85E).tav MJ_3983(85E_vs_95-2).tav 2nd_set_MJ_3983(85E_vs_952)_MJ_4008(95-2_vs_85E).tav MJ_4008(95-2_vs_85E).tav MJ_7978(85E_vs_95-2).tav 3rd_set_MJ_7978(85E_vs_952)_MJ_7937(95-2_vs_85E).tav MJ_7937(95-2_vs_85E).tav