Supplementary Texts (doc 59K)
advertisement
")
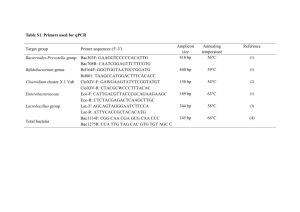
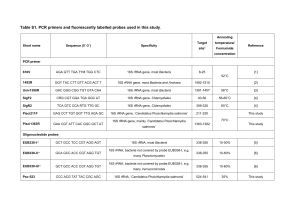
1 Supplementary Texts, Imachi et al. 2 3 Supplementary Text 1, 4 The reason for why these organic substances were used for cultivation of subseafloor 5 methanogenic community. The substrates for the enrichment of subseafloor methanogenic 6 microbial communities are important. In this study, we provided glucose, yeast extract, 7 acetate and propionate as potential energy and carbon sources for the enrichment in the DHS 8 reactor. Other than acetate, methanogens cannot directly use these substances, but these 9 substrates are more realistic energy and carbon sources for the in situ subseafloor sedimentary 10 habitat. Most of the methanogens can grow on H2 and carbon dioxide as methanogenic 11 substrates (Liu and Whitman, 2008). In many natural anaerobic habitats, including the 12 subseafloor sediments, methanogens should thrive by receiving H2 that is provided by 13 heterotrophic H2-producing bacteria, which catalyze the oxidation of a variety of organic 14 substances (Liu and Whitman, 2008; Stams and Plugge, 2009). The methanogens utilize the 15 H2 produced by these heterotrophic bacteria, and in return, the bacteria benefit from the 16 removal of excess H2 that would otherwise inhibit their growth. This relationship is 17 commonly referred to as interspecies H2 transfer (Schink, 1997; Stams and Plugge, 2009). In 18 syntrophic associations of methanogens and heterotrophic bacteria via interspecies H2 transfer, 19 H2 is continuously provided at a low concentration from heterotrophic bacteria to H2-utilizing 20 methanogens (less than 30 Pa in the case of propionate) (Imachi et al., 2000; Sakai et al., 21 2007). Furthermore, organic substances, particularly fatty acids, are generally converted to H2 22 at a slow rate by heterotrophic bacteria because bacteria that live in syntrophy with 23 H2-methanogens are generally exhibit slow growth rates (approximate 5 day doubling time in 24 the case of syntrophic propionate-oxidizing bacteria [Harmsen et al., 1998; Imachi et al., 25 2007]). Therefore, we focused on interspecies H2 transfer, and we have proposed a new 1 1 method for cultivating H2-utilizing methanogens; the co-culture method (Sakai et al., 2007; 2 Sakai et al., 2009). Previously, we successfully cultivated phylogenetically diverse 3 methanogens in serum vials containing a defined medium supplemented with ethanol or fatty 4 acids as indirect precursor substrates that are converted to H2 by heterotrophic bacteria, and 5 we eventually isolated novel methanogens in pure culture (Imachi et al., 2008; Sakai et al., 6 2008). Therefore we assumed that the organic substances used in the current study might also 7 be one of the appropriate substrates for the cultivation of marine subsurface methanogens that 8 have adapted well to an extremely slow H2 flux and might form syntrophic associations with 9 heterotrophic H2-producing bacteria in their natural setting. 10 References 11 Harmsen HJM, Van Kuijk BLM, Plugge CM, Akkermans ADL, De Vos WM, Stams AJM. 12 (1998). Syntrophobacter fumaroxidans sp. nov., a syntrophic propionate-degrading 13 sulfate-reducing bacterium. Int J Syst Bacteriol 48: 1383-1387. 14 Imachi H, Sakai S, Ohashi A, Harada H, Hanada S, Kamagata Y et al. (2007). Pelotomaculum 15 propionicicum sp. nov., an anaerobic, mesophilic, obligately syntrophic, propionate-oxidizing 16 bacterium. Int J Syst Evol Microbiol 57: 1487-1492. 17 Imachi H, Sakai S, Sekiguchi Y, Hanada S, Kamagata Y, Ohashi A et al. (2008). Methanolinea 18 tarda gen. nov., sp. nov., a methane-producing archaeon isolated from a methanogenic 19 digester sludge. Int J Syst Evol Microbiol 58: 294-301. 20 Imachi H, Sekiguchi Y, Kamagata Y, Ohashi A, Harada H. (2000). Cultivation and in situ 21 detection of a thermophilic bacterium capable of oxidizing propionate in syntrophic 22 association with hydrogenotrophic methanogens in a thermophilic methanogenic granular 23 sludge. Appl Environ Microbiol 66: 3608-3615. 24 Liu Y, Whitman WB. (2008). Metabolic, phylogenetic, and ecological diversity of the 25 methanogenic Archaea. Ann N Y Acad Sci 1125: 171-189. 2 1 Sakai S, Imachi H, Hanada S, Ohashi A, Harada H, Kamagata Y. (2008). Methanocella 2 paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage 3 'Rice Cluster I', and proposal of the new archaeal order Methanocellales ord. nov. Int J Syst 4 Evol Microbiol 58: 929-936. 5 Sakai S, Imachi H, Sekiguchi Y, Ohashi A, Harada H, Kamagata Y. (2007). Isolation of key 6 methanogens for global methane emission from rice paddy fields: a novel isolate affiliated 7 with the clone cluster Rice Cluster I. Appl Environ Microbiol 73: 4326-4331. 8 Sakai S, Imachi H, Sekiguchi Y, Tseng I-C, Ohashi A, Harada H et al. (2009). Cultivation of 9 methanogens under low-hydrogen conditions by using the coculture method. Appl Environ 10 Microbiol 75: 4892-4896. 11 Schink B. (1997). Energetics of syntrophic cooperation in methanogenic degradation. 12 Microbiol Mol Biol Rev 61: 262-280. 13 Stams AJM, Plugge CM. (2009). Electron transfer in syntrophic communities of anaerobic 14 bacteria and archaea. Nat Rev Microbiol 7: 568-577. 15 16 Supplementary Text 2, 17 Chemical analyses. The temperature, pH and oxidation-reduction potential (ORP) of effluent 18 seawater were measured using an InPro3250 pH and redox sensor (Mettler Toledo). 19 Concentrations of acetate, propionate and other organic acids such as succinate, malate, 20 fumarate, butyrate and lactate were measured by HPLC using a Shim-pack SCR-102H 21 column (Shimadzu; mobile phase, 4 mM p-toluenesulfonic acid; column temperature, 45°C). 22 Glucose and ethanol concentrations were determined by HPLC using an SCR101-H column 23 (Shimadzu; eluent, H2O; column temperature, 60°C) and a refractive index detector 24 (Shimadzu RID-10A). The TOC was measured using a TOC analyzer (TNC-6000, Toray 25 Eng.) according to the Japanese Industrial Standards (JIS K0102 22.1). Methane 3 1 concentration was determined by gas chromatography (GC) (GC3200G, GL Science) with a 2 thermal conductivity detector. Measurement of dissolved methane in effluent seawater was 3 performed as previously described (Hatamoto et al., 2010). The stable carbon isotope 4 compositions of CH4 and CO2 in the sampled gas phase were analyzed by Taiyo-Nissan Co. 5 Ltd. using a SerCon ANCA-ORCHID GC isotope ratio mass spectrometer (SerCon). The 6 concentration of monomethylamine was measured by HPLC using a Mightysil RP-18 PA 7 column (Kanto Chemical; eluent, acetonitrile/H2O) and a UV detector (Waters 996 8 Photodiode array detector) at 340 nm, after induction by 2,4,6-trinitrobenzene sulfonic acid. 9 Methanol was analyzed by GC-mass spectrometry (JEOL JMS-AM20) using a DB-WAX 10 column (Agilent Technologies). Trimethylamine was analyzed by GC (Shimadzu GC-2014) 11 using a Chromosorb W column (Shinwa Chemical Industries) and a flame photometric 12 detector. All effluent seawater samples were filtered with a 0.22 µm pore-size 13 polyethersulfone filter unit (Millipore) immediately after sampling and stored at 4°C until 14 measurements. 15 Nucleic acid extraction, PCR and cloning. DNA extraction and PCR amplification were 16 performed as described previously (Miyashita et al., 2009). For PCR amplification, we used 17 the primer pairs Arch21f (DeLong, 1992)/Ar912r (Miyashita et al., 2009) and EUB338F* 18 (Amann et al., 1990; Daims et al., 1999)/1492R (Weisburg et al., 1991) for the construction 19 of 16S rRNA gene-based archaeal and bacterial clone libraries, respectively. For construction 20 of archaeal 16S rRNA gene-based clone libraries from enrichment cultures, Acrh21f-Mvb 21 (5’-TTC TGT TTG ATC CTG GCA GA-3’) was used together with Arch21f as the forward 22 primer (in equal concentrations), in order to cover Methanobrevibacter species 16S rRNA 23 gene sequences (see dialed explanation Supplementary Text 3). We also used primers 24 Arch21F (or Arch21F-Mvb)/1492R or 8f (Weisburg et al., 1991)/1492R to obtain the nearly 25 full-length 16S rRNA gene from archaeal and bacterial isolates, respectively. For PCR 4 1 amplification of the mcrA gene, we used primers Luton-mcrA (Luton et al., 2002) and 2 ME1/ME3 (Hales et al., 1996) for the clone analysis and methanogenic isolates, respectively. 3 The PCR conditions were the following: initial denaturation at 95°C for 10 s, followed by 20 4 to 35 cycles of denaturation at 95°C for 5 s, annealing at 50°C for 30 s, and extension at 72°C 5 for 90 s. To reduce possible bias caused by PCR amplification, we used PCR products 6 obtained at minimized PCR cycle numbers ranging from 20 to 35 cycles at five-cycle 7 intervals. Clone library construction and sequencing were performed as described previously 8 (Miyashita et al., 2009). 9 Total RNA extraction from the enrichment samples was performed immediately after 10 sampling from the DHS reactor using the method described previously (Sekiguchi et al., 11 2005). The remaining DNA was digested with RNase-free DNase I (Promega). The absence 12 of contaminating genomic DNA in the RNA extract was confirmed by PCR using the primer 13 pairs EUB338F*/907r (Lane, 1991) and Arch21f/Ar912 with 35 PCR cycles. The 14 concentration of RNA was quantified spectrophotometrically with a Quanti-iT RNA assay kit 15 (Invitrogen). Reverse transcription (RT)-PCR was performed with a commercial RT-PCR kit 16 (SuperScript III One-Step RT-PCR System with Platinum Taq DNA polymerase, Invitrogen) 17 according to the manufacturer’s instructions. The same primer sets were used for the RT-PCR 18 of 16S rRNA and mcrA mRNA as were used for the DNA-based clone analyses described 19 above. The subsequent procedures were also the same as used for the DNA-based clone 20 analyses described above. 21 FISH. The samples were fixed with 2% paraformaldehyde in anaerobic synthesis seawater 22 excluding organic substances for 12 h at 4°C and stored in 50% ethanol with 23 phosphate-buffered saline (PBS; 130 mM NaCl, 10.8 mM Na2HPO4, 4.2 mM NaH2PO4 [pH 24 7.2]) at -20°C. For FISH detection, the fixed samples were diluted in 1 x PBS and sonicated to 25 disperse cells at 20 W for 1 min on ice using an ultrasonic homogenizer (Model UH-50, SMT 5 1 Co. Ltd.). Each sample (approximately 0.5 g dry wt) was transferred to a 1.5 ml tube. Then, 2 FISH was performed according to a previous report (Sekiguchi et al., 1998), modified by the 3 addition of 1% blocking reagent (w/v, Roche Diagnostics) to the hybridization buffer. 4 Catalyzed reporter deposition (CARD)-FISH with the horseradish peroxidase 5 (HRP)-labeled ARC915 probe (Thermo Electron Biopolymer) was performed based on a 6 method described previously (Kubota et al., 2006; Pernthaler and Amann, 2004). For 7 CARD-FISH experiments, the fixed samples were embedded in a MetaPhor low melting point 8 agar (Cambrex), and the following pretreatments for the incremental penetration of the 9 HRP-labeled probe into fixed archaeal cells were performed: (i) lysozyme treatment (10 10 mg/ml in 100 mM Tris-HCl [pH 7.5] and 50 mM EDTA [pH 8.0]) at 37°C for 30 min; (ii) 11 proteinase K treatment (20 µg/ml in 100 mM Tris-HCl [pH 7.5] and 50 mM EDTA [pH 8.0]) 12 at 37°C for 1 hour; and (iii) 0.01 M HCl for 10 min at room temperature. To inactivate 13 endogenous peroxidase, the cell samples were incubated with H2O2 solution (final 14 concentration, 0.3% [v/v] in methanol) for 10 min at room temperature. As a negative control, 15 sediment samples were hybridized with an HRP-labeled nonsense-probe, NON338 (Manz et 16 al., 1992), which did not produce a CARD-FISH signal in the samples. 17 References 18 Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. (1990). Combination 19 of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed 20 microbial populations. Appl Environ Microbiol 56: 1919-1925. 21 Daims H, Brühl A, Amann R, Schleifer KH, Wagner M. (1999). The domain-specific probe 22 EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a 23 more comprehensive probe set. Syst Appl Microbiol 22: 434-444. 24 DeLong EF. (1992). Archaea in coastal marine environments. Proc Natl Acad Sci USA 89: 25 5685-5689. 6 1 Hales BA, Edwards C, Ritchie DA, Hall G, Pickup RW, Saunders JR. (1996). Isolation and 2 identification of methanogen-specific DNA from blanket bog peat by PCR amplification and 3 sequence analysis. Appl Environ Microbiol 62: 668-675. 4 Hatamoto M, Yamamoto H, Kindaichi T, Ozaki N, Ohashi A. (2010). Biological oxidation of 5 dissolved methane in effluents from anaerobic reactors using a down-flow hanging sponge 6 reactor. Water Res 44: 1409-1418. 7 Kubota K, Ohashi A, Imachi H, Harada H. (2006). Visualization of mcr mRNA in a 8 methanogen by fluorescence in situ hybridization with an oligonucleotide probe and two-pass 9 tyramide signal amplification (two-pass TSA-FISH). J Microbiol Methods 66: 521-528. 10 Lane DJ. (1991). 16S/23S rRNA sequencing. In: Stackebrandt E and Goodfellow M (eds). 11 Nucleic Acid Techniques in Bacterial Systematics. John Wiley & Sons: Chichester, United 12 Kingdom, pp 115-175. 13 Luton PE, Wayne JM, Sharp RJ, Riley PW. (2002). The mcrA gene as an alternative to 16S 14 rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology 148: 15 3521-3530. 16 Manz W, Amann R, Ludwig W, Wagner M, Schleifer K-H. (1992). Phylogenetic 17 oligodeoxynucleotide probes for the major subclasses of Proteobacteria: problems and 18 solutions. Syst Appl Microbiol 15: 593-600. 19 Miyashita A, Mochimaru H, Kazama H, Ohashi A, Yamaguchi T, Nunoura T et al. (2009). 20 Development of 16S rRNA gene-targeted primers for detection of archaeal anaerobic 21 methanotrophs (ANMEs). FEMS Microbiol Lett 297: 31-37. 22 Pernthaler A, Amann R. (2004). Simultaneous fluorescence in situ hybridization of mRNA 23 and rRNA in environmental bacteria. Appl Environ Microbiol 70: 5426-5433. 24 Sekiguchi Y, Kamagata Y, Syutsubo K, Ohashi A, Harada H, Nakamura K. (1998). 25 Phylogenetic diversity of mesophilic and thermophilic granular sludges determined by 16S 7 1 rRNA gene analysis. Microbiology 144: 2655-2665. 2 Sekiguchi Y, Uyeno Y, Sunaga A, Yoshida H, Kamagata Y. (2005). Sequence-specific 3 cleavage of 16S rRNA for rapid and quantitative detection of particular groups of anaerobes 4 in bioreactors. Wat Sci Technol 52: 107-113. 5 Weisburg WG, Barns SM, Pelletier DA, Lane DJ. (1991). 16S ribosomal DNA amplification 6 for phylogenetic study. J Bacteriol 173: 697-703. 7 8 Supplementary Text 3. 9 An explanation for the use of archaral primer Arch21f-Mvb. In our clone analyses for 10 enrichment samples from the DHS reactor, an mcrA phylotype related to the genus 11 Methanobrevibacter (357D_mcrA1) was detected in the clone libraries, while a 16S rRNA 12 gene phylotype belonging to Methanobrevibacter was not detected. During the clone analysis, 13 we realized that the 16S rRNA gene sequence of Methanobrevibacter probably had 14 mismatches with the Arch21f primer because the complete genome sequence of 15 Methanobrevibacter smithii (GenBank accession number NC_009515) contains 4 mismatches 16 with the Arch21f primer. This primer mismatch presumably caused the Methanobrevibacter 17 phylotype to be missed in the 16S rRNA gene clone libraries. Indeed, after addition of the 18 modified primer Arch21f-Mvb to the PCR amplification, a 16S rRNA phylotype that was very 19 closely related to Methanobrevibacter members was successfully detected from the batch-type 20 cultures (Table S8). Thus, we should pay close attention to select or update/revise PCR 21 primers when archaeal diversity is estimated, because some archaeal PCR primers have high 22 mismatch frequencies in particular marine subsurface archaeal lineages (Teske and Sørensen, 23 2008). 24 Reference 25 Teske A, Sørensen KB. (2008). Uncultured archaea in deep marine subsurface sediments: 8 1 have we caught them all? ISME J 2: 3-18. An explanation for the use of archaral primer 2 Arch21f-Mvb. 9