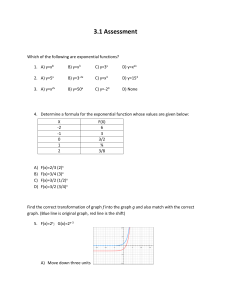
THE UNIVERSITY OF NORTH CAROLINA AT
advertisement

THE UNIVERSITY OF NORTH CAROLINA AT CHAPEL HILL BIOLOGY 421L and 422L LABORATORY MANUAL Fall, 2013 DR. MATTHYSSE Biology 421L and 422L Laboratory Experiments Fall, 2013 Laboratory objective – Both labs: To learn some of the major techniques and procedures used in working with bacteria. Although the results you will obtain in these experiments are standard and easily verifiable in published papers and texts, the experiments are designed to provide you with experience in a variety of the most commonly used techniques of bacteriology. In a working microbiology laboratory if you wished to use a new technique you would check whether you were doing the technique correctly by using it in a standard well-documented experiment as you are doing here. 421L (two credit lab) only: To gain experience with the approaches and methods used in conducting scientific experiments. You will design and carry out a real experiment providing new information to the scientific community. Schedule for 421L (2 credit lab) Date Experiment Hand In Aug. 27 & *1) Sterile Technique 1) Plates Sept. 3 (S) Viable cell count Plate streaking Sept. 10 (L) *2) Growth Curve 2) Report (graph & 2 pages) OD growth curve Gram stain Separating individual colonies 2)Hand in plates Sept. 17 (L) *3) β-galactosidase Induction 3) Report (graph, table & 2 pages) Sept. 24 *4) Isolation of Bacteria from Nature --*5) Transposon mutagenesis Part 1 Oct. 1 *4) Isolation of bacteria from nature 4) Report (2-4 pages) & Chart. *5) Transposon mutagenesis Part 2 Due after experiment completed Oct. 8 *5) Transposon mutagenesis Part 2 5) Report (2 pages) 6) Biochemical Pathways Plate of Mutants 8) Part1 Begin planning your experiment 6) Report (2 pages) Oct. 15 8) Part 1 Plan and make materials for your experiment Oct. 22 7) One-Step Phage Growth 7) Report (graph, OD growth curve, 8) Continue working on your experiment plaque counts) Oct. 29 (L) 8) Part 2 Transposon mutagenesis: preparation 8) Report should be in form of a of the plasmid and introduction of the journal paper (one report per group) transposon due after Thanksgiving Nov. 5 (L) 8) Part 3 Screen and characterize mutants. Nov. 12 8) Part 4 Continue characterizing mutants and prepare stocks of the mutants Nov. 19 (S) *Completion of all experiments *Clean up lab *Meet to discuss lab Dec. 3 (S) *Hand in all lab reports *Laboratory final exam (S) short lab You should be done by 3 PM. (L) long lab this lab may last after 4 PM: if this is a problem for you, make arrangements with your TA before the lab. * Labs which will be done by 422L Please note that you will need to come in the next day between 9AM and noon and 2 and 5PM to check your plates and transfer your cultures. The lab will be open 9 to 5 weekdays. However, you should avoid coming in during the first hour of any other lab section since this may disrupt that section. Date (all labs begin at 1 PM) Aug. 28 & Sept. 4 (S) Sept. 11 (L) Sept. 18 (L) Sept. 25 Oct. 2 Oct. 9 (S) Oct. 23 (S) Dec. 4 (S) Schedule for 422L (one credit lab) Experiment 1) Sterile technique 2) Growth curve, Gram stain, pure cultures 3) β-galactosidase induction 4) Isolation of bacteria from nature 5) Transposon mutagenesis part 1 4) Isolation of bacteria from nature 5) Transposon mutagenesis Part 2 5) Transposon mutagenesis finish Clean up lab Meet to discuss lab Laboratory final exam (S) short lab You should be done by 3 PM. (L) long lab this lab may last after 4 PM: if this is a problem for you, make arrangements with your TA before the lab. Please note that you will need to come in the next day between 9 AM and 5 PM to check your plates and transfer your cultures. The lab will be open 9 to 5 weekdays. However, you should avoid coming in during the first hour of any other lab section since this may disrupt that section. General Information for Biology 421L and 422L Before each class meeting, read the directions and references for that laboratory. This will help you to use your time to the best advantage. All the laboratory experiments have preliminary worksheets included in the manual. The pre-lab worksheets are designed to require you to read the lab to complete them. These are to be completed and turned in before you begin the lab. Laboratory is scheduled for only 3 hours on Tuesday or Wednesday afternoons. The remaining hours of laboratory will be the time the next day when you come in to examine plates, test cultures, etc. You may do this on your own. If you wish help, schedule an appointment with the teaching assistants or Dr. Matthysse generally goes to the lab briefly after lecture and will help you then. If you are not satisfied with your results on a particular experiment, some (but not all) experiments can be repeated. You should arrange a time to do this with the teaching assistants. The week following most of the laboratory experiments a brief report will be due. All reports must be printed (except diagrams or graphs which may be hand drawn, use the graph paper in the back of this manual) and handed directly to the TA concerned. A rubric for each of the lab reports will be distributed by your TA before the report is due. In addition, for some experiments you may need to hand in plates or cultures. Some laboratory exercises will also include problems for you to work. The laboratory grade will be based on the laboratory reports and plates handed in, the lab final exam, and the teaching assistant’s evaluation of your microbiology laboratory skills. Reports will be graded on: 1. Clarity and organization 2. Scientific correctness of results 3. Completeness - all questions answered. All results present. All calculations made. All procedures described (if you used the procedures in the directions, it is adequate to state this). 4. All reports should contain your data from the experiment in a table or other easily readable form. If for some reason you were unable to obtain data, consult with your TA about what to do. If you use someone else's data or invented data, this must be clearly stated. In the case in which you use data from another student, they must sign the report to show that they consent to your use of their data. While it may be unavoidable and no fault of yours that you have to use someone else’s data, this should be a rare occurrence. If you are having trouble getting good data, consult with the teaching assistants or Dr. Matthysse regarding your lab technique. Poor lab technique will affect your grade adversely. 5. Late lab reports will lose 1 point for each week day they are overdue. No lab report will be accepted after Dec. 1. The final examination will consist of protocols and data similar to those from the experiments done in class. It will last 1 hour. Grades will be calculated with each laboratory from number 2 through 8 (2 credit lab) or 2 through 5 (1 credit lab) counting 10 points except the laboratory on isolation of bacteria from nature which will count 20 points and laboratory 8 which will count 40 points. The final will count 30 points and an evaluation of your work in the laboratory by the TA will count 40 points. The TA evaluation will be based on their assessment of your lab skills and technique and understanding of the experiments. Laboratory Safety Before you come to the laboratory. Good sense and OSHA safety regulations require that you wear sensible clothing which will protect you from chemical spills and closed shoes. You should not have long floppy sleeves or long hair which may hang into a burner flame and catch on fire. Fasten your hair back securely. Anyone with an impaired or suppressed immune system should not enter a microbiology laboratory. General Laboratory Safety. You may not eat, drink or smoke in the laboratory. You should store your coat and back pack out of the way where no one will trip on them and they will not become contaminated with microorganisms. If you leave them out and live cultures are spilled on them they will have to be disinfected. Precautions in working with microorganisms. You should treat all cultures of microorganisms as if they contained human pathogens since you do not know what contaminants you may have grown. Never mouth pipette cultures. Use a pi pump or rubber bulb. Wash the area where you will work with disinfectant before you begin work. If you spill a drop of culture, kill it with disinfectant or alcohol at once. Place all pipettes tip down in disinfectant immediately after use. Never vortex an open culture or carry out any other procedure which could produce an aerosol of a microorganism. Kill all liquid cultures, dilution tubes of microorganisms, etc. with disinfectant when you are done with them. Do not leave glassware carrying live organisms lying around. When you have used a loop or spreader to transfer or spread bacteria, kill the remaining organisms (in the flame or alcohol) before you place the loop or spreader on the bench top. At the end of the experiment wash the bench top with disinfectant. Wash your hands before you leave the lab. Bacteriophage are not killed by disinfectant; use NaOH or Clorox bleach instead to kill them. Precautions in working with chemicals. Never mouth pipette chemicals. Use a pi pump or a rubber bulb. If working with organic chemicals wear safety glasses (in your drawer). Wear gloves to work with phenol or other caustic agents. Never engage in procedures which could cause caustic or organic solvents to splash such as vigorous mixing or shaking of open containers. Never leave unlabeled chemical solutions sitting around. If you wish to keep them, close them properly in a screw cap container and label them. If you wish to discard them, dispose of them in the sink or in marked containers and rinse the glassware before leaving it to be washed. Never put unrinsed glassware in the wash. The chemicals on it could react with those on other unwashed glassware to form hazardous compounds. Ultraviolet light. UV light is harmful to your skin and eyes. Always wear a face shield, gloves, and long sleeves when working with UV light sources. Cover any part of the source you do not need. Limit your exposure time to that which is necessary. Long wavelength UV is less dangerous than short wavelength UV. Burners, alcohol fires, and paraffin fires. Remember fire is dangerous. Before you turn on and light a Bunsen burner, check that there are not open containers of flammable liquids nearby. Also check that there are no containers such as squirt bottles which may respond to the heat from the burner by releasing liquid. Similar checks should be made before turning on a hot plate. Ethanol catches on fire quite easily. Fortunately, such fires are not very hot. If you light alcohol in a Petri dish or beaker on fire, simply cover it with a glass lid to suffocate the flame due to lack of oxygen. Paraffin if is heated too hot on a hot plate it will burst into flames. Cover it to suffocate the flame. Turn OFF the hot plate. If you should somehow catch your hair or clothes on fire, put out the fire by smothering it. Do not run. Instead roll on the floor to put out the flames. Do not attempt to smother a fire with coats, etc. unless you know that the material is not highly flammable. Many synthetic fibers burn easily or even explode if exposed to fire. General Directions for an Afternoon in the Laboratory Before you come to the lab read the lab manual carefully, do the pre-lab worksheet, and make a flow chart of what you will do in the lab including tables to record your data. When you come to the lab1. Hang your coat and store your back pack out of the way. Do not leave them out. If they become contaminated with bacterial cultures they will need to be disinfected. 2. Wash the area where you will work with disinfectant. 3. Lay out the materials and tools you will need for the experiment. Get out your lab manual and your flow chart. Hand in your pre-lab exercise. 4. Listen to the pre-lab directions and explanations from your TA. 5. Do the experiment. Always notice how your cultures and other materials appear. If anything does not seem right check with your TA. 6. When you have finished the experiment, place all cultures (carefully labeled) which need to grow in the proper location- the 37o incubator or in your cupboard for room temperature or in front of the light for photosynthetics. 7. When you are done, kill all cultures and other contaminated solutions, etc. with disinfectants. Kill organisms in side-arm flasks with ethanol. Rinse all glassware and either load it for the dishwasher or in the case of side-arm flasks wash it yourself and hang it up to dry. 8. Clean your work area. Throw away used disposable pipettes, etc. after they have been treated with disinfectant. 9. Check with your TA about when to check your cultures. 10. Wash your hands before you leave the lab. When you check your culturesa. Once again, first store your coat and back pack away. Even if there are few people around materials could be spilled on your possessions. b. Get out your lab notebook and turn to the data tables you prepared for this lab. c. Get your cultures and record your data. d. Kill all cultures with disinfectants or put them in autoclave bags. Wash any dishes. Wash your lab bench with disinfectant and leave it clean. e. If you cultures look strange or unexpected, store then in your room temperature cupboard or the refrigerator and ask your TA or Dr. Matthysse for help. Do not carry your cultures out of the lab to ask for help. Instead ask some one to come to the lab and look at them. f. Wash your hands before you leave the lab. Biology 422L Outline of a Typical Lab Report 1. Objective. One or two sentences in your own words. 2. Notes on procedures. Deviations from the lab manual. Anything unexpected that happened. Maximum length: one paragraph. 3. Results. Show your original data in a table. If appropriate also show data as a graph. Show your calculations including the equations you used. Organize everything. Be concise. Draw any conclusions that seem valid. State your reservations, if any, about your conclusions clearly. 4. Answer any questions asked in the lab manual or by your TA. 5. Keep your report concise. Extra verbiage will not get you extra points and may actually result in a loss of points. Name___________________________ Pre-Laboratory Exercise Laboratory 1 1. What would you see the next day on a nutrient agar plate inoculated with E. coli, if you failed to use sterile technique while working with the plate? 2. If you perform a viable cell count and obtain 32 colonies on a plate on which you put 0.1 ml of a 10--8 dilution of the original bacterial culture, how many bacteria per ml were there in the original culture? Biology 422L Laboratory 1 Sterile Technique Objective: To learn sterile technique. Reference: Slonczewski and Foster Chapter 4 Please read references for laboratory experiments before coming to the laboratory so that you can make the most use of the limited time available to do the experiments. The ability to isolate and maintain a pure culture of a micro-organism is basic to all laboratory work in microbiology. Your instructor will demonstrate sterile technique which is designed to eliminate contamination and to allow the cultivation of a pure stain. 1) To convince yourself that contamination from the air is a real possibility leave an open Petri plate of nutrient agar on the laboratory bench for the afternoon. Then incubate it overnight at 37oC. 2) You will be provided with an overnight culture of Escherichia coli. Those students who have no previous experience with sterile technique may wish to practice it before beginning the experiment. Using an inoculating loop and the technique demonstrated by your instructor streak 2 or 3 plates of sterile nutrient agar. Label all Petri plates with your name, the contents of the plate, and the date on the bottom side. Incubate plates at 37oC bottom side up so that any water which condenses in the plate will not run onto your colonies. For E. coli overnight incubation at 37oC is usually sufficient to give good growth. When the bacteria have grown, Petri plates can be stored in the refrigerator. NOTE - You will use this technique many times during the semester. 3) The number of viable cells per ml in an overnight culture is very large. To determine how many cells there are it is necessary to dilute them. Usually ten-fold dilutions in sterile buffer are used. Take tubes containing 0.9 ml of sterile buffer and using 0.1 ml of the overnight culture make a 1 to 10 dilution. Mix the dilution well by vortexing it. Then using a fresh sterile pipette transfer 0.1 ml of the dilution to a new tube of sterile buffer. Repeat until you have reached a dilution of 10-8. Then pipette 0.1 ml of each dilution form 10-5 to 10-8 onto nutrient agar plates. Spread the bacterial suspension on the plate with a bent glass rod which is sterilized by dipping it in alcohol and burning off the alcohol. This technique will be demonstrated for you. When the plates are spread, let them sit right-side up for a few (about 10) minutes to dry. Then incubate them overnight at 37oC, bottom side up. Sometime the next day remove your plates from the incubator and either count the colonies per plate then or store the plates in the cold and count them later. NOTE - You will use this technique many times during the semester. See separate page for sample calculations. Report: It is unnecessary to submit a written laboratory report for this exercise. Instead, each student must submit an uncontaminated streaked plate with some individual colonies and a set of uncontaminated dilution plates. If the plates you prepare during the laboratory period appear to be contaminated or otherwise unsatisfactory when you look at them the next day, then you should prepare new dilutions from a fresh culture and inoculate fresh plates. Repeat this process until you obtain a satisfactory streaked plate and set of dilution plates. Biology 422L General Bacteriological Technique and Sample Calculations A) Streaking plates - use this technique to obtain pure cultures of organisms from a mixed sample. 1. Flame your loop. 2. Cool it by touching it to a sterile uninoculated place on the agar plate. 3. Take a loopful of mixed culture or pick a colony from a plate. 4. Streak back and forth across the top 1/3 of your plate. Press lightly as if making a watercolor painting. 5. Flame your loop. 6. Cool it by touching it to a sterile uninoculated place on the agar plate. 7. Spread the bacteria out further by running your loop through the inoculated part of the plate once and then streaking about 1/2 of the uninoculated part of the plate. Do this at a right angle to the original streaks. 8. Flame your loop. 9. Cool it by touching to a sterile uninoculated place on the agar plate. 10. Spread the bacteria out in a further dilution by running your loop through the second set of streaks (#7) at a right angle once and then streaking the remainder of the plate at a right angle to the streaks made in #7. You may have to adjust the positions of the plate and your eyes to be able to see the streaks you have made. 11. Turn the plate over. Label it on the bottom and incubate it at the appropriate temperature. B) Viable cell counts - Use this technique to determine the number of viable cells per ml of liquid. 1. Place 0.9 ml of sterile solution (usually 0.9% NaCl) in each of several sterile tubes (the number depends on how many dilutions you wish to make). Label the tubes 1, 2, 3 etc. 2. Take 0.1 ml of your liquid culture and add it to tube #1 using sterile technique. Vortex tube #1. This is a 1/10 (10-1) dilution. 3. Take 0.1 ml of tube #1 and add it to tube #2. Vortex tube #2. This is a 1/100 (10-2) dilution. 4. Repeat Step 3 until you come to the end of your tubes. 5. Take a set of agar plates and label them on the bottom with your name, date, culture, and dilution # (e.g. 5 or 6 or 7). 6. Take the appropriate tube, vortex it, and pipette 0.1 ml onto the plate of the same number. 7. Dip your glass spreader into alcohol to sterilize it. Burn off the alcohol using your burner flame. (Do not allow the glass to become hot). Using the bent end of the glass spread the 0.1 ml of liquid over the plate going back and forth in one direction across the plate. Then turn the plate 90o and spread back and forth across the plate again. Repeat this until the plate is evenly covered. When you are done dip your glass spreader in alcohol to kill any bacteria remaining on it. 8. Allow your plate to dry about 15 min. Then invert it and incubate it at the appropriate temperature. 9. Repeat 6, 7, and 8 for each plate to be spread. C) Sample calculations for a viable cell count. 1. If 0.1 ml of tube 1 (10-1 dilution) is spread on a plate and 57 colonies grow then there were 57 x 10 colonies in 0.1 ml of the original culture or 57 x 10 x 10 colonies in 1.0 ml of the original culture. The viable cell count is 5.7 x 103 bacteria per ml. 2. If 0.1 ml of tube 7 (10-7 dilution) was spread and 38 colonies grew then there were 38 x 107 bacteria in 0.1 ml or 38 x 108 in 1.0 ml. The viable cell count is 3.8 x 109. 3. If 0.1 ml of tubes 4, 5, 6, and 7 (10-4, 10-5, 10-6, 10-7 dilutions) were plated and about 2,000, 280, 25, and 4 colonies grew. The viable cell counts measured from each are 2 x108, 2.8 x 108, 2.5 x 108, and 4 x 108. The statistical sampling error is the square root of the number of colonies observed so these numbers become 2 + .04, 2.8 + 0.17, 2.5 + 0.5, and 4 + 2 x 108. Thus you would estimate the viable cell count from these numbers at 2.8 + 0.2 x 108 since the 280 number is probably the most reliable. (It is difficult to count 2,000 colonies accurately and the other numbers are too small for statistical accuracy). Name____________________________ Pre-Laboratory Exercise Laboratory 2 1. How would you obtain a pure culture of a microorganism from a soil sample containing many different species of bacteria? 2. The following data was obtained during a growth curve: Time (min) OD of culture 0 0.05 10 0.05 20 0.07 30 0.10 40 0.13 50 0.20 60 0.23 70 0.24 80 0.24 Plot the data using a linear scale for time and a logarithmic scale for the culture OD. (There is semi-log graph paper in the back of this manual). If you are unfamiliar with semi-log plots the following information may help you. The horizontal scale on this type of graph paper is the familiar linear scale; but the vertical scale is logarithmic. Thus numbers which increase exponentially if plotted on this scale will appear linear. Try this using the following series of numbers (horizontal numbers are given first and the vertical numbers to go with them are given second): 0,1; 1,2; 2,4; 3,8; 4,16; 5,32. These data are graphed on the next page. When using semi-log paper you must choose the particular range you wish to use. You could have the bottom cycle from 0.01 to 0.1, the next cycle from 0.1 to 1.0 and the next cycle from 1 to 10, etc. or whatever you choose. On your bacterial growth curve, identify lag, log, and stationary phase. What is the doubling time of the culture in log phase? Biology 422L Laboratory 2 Bacteriological Techniques Objectives: 1) To continue to practice sterile technique until it becomes easy for you to do. 2) To observe the growth of bacteria and see how to test the effect of a substance on bacterial growth. 3) To observe bacteria in the light microscope and learn how to do a Gram stain. 4) To use the technique of streaking plates which you learned in Experiment 1 to separate a mixed culture of bacteria. The ability to do this is often needed in a microbiology laboratory. References: Slonczewski and Foster, Chapter 4 Bacteria can be grown in either liquid or solid medium. Liquid cultures provide a relatively uniform suspension of growing cells, particularly if the cells are harvested during exponential growth. On solid medium the cells, if they are well dispersed and not too crowded, will each give rise to a colony of 106 to 108 cells all of which are the progeny of a single cell (a clone). 1) Growth curve. The growth of cells in liquid medium can be followed by measuring the optical density of the cell suspension. Sterile side arm flasks and tubes of sterile medium will be provided. Pipette 5 ml of medium into a flask. Half the students should use minimal medium; the other students should use nutrient broth. Different students should add 0.5 ml of either 1% or 0.1% glucose solution to the minimal medium - to give a final concentration of 0.1% or 0.01% glucose. Add 0.5 ml of an overnight culture of E. coli in Nutrient Broth to each flask (use 0.1 ml if the E. coli were grown in Luria broth). Label the flask with your name and make sure the plug fits tightly. Read and record the optical densities of each solution at 550 nM (instructions will be given for the use of the spectrophotometers). Subtract the optical density of the medium alone from your readings (this can be done by using a blank containing the same medium). Place the flasks on a shaker in a 37o water bath and follow the growth by reading the optical density roughly every 15 min (record actual time of readings). (See figure for the correct way to place flasks on the shaker). At the end of the experiment place plugs in the container provided. DO NOT THROW THEM AWAY. Record the data from the whole class. After 1-1/2 hrs of growth a few students using nutrient broth cultures may add streptomycin, ampicillin, lysol or any "bactericidal" agent purchased in a drug store to their growing cultures. These students should determine the OD and the number of viable cells in their cultures just before the addition is made and then again 1/2 hr later. When the cultures have all reached stationary phase (or after 2 hrs) determine the number of viable cells per ml in your culture flask using the techniques from laboratory No. 1. Plate dilutions of 10-4 to 10-6. After the addition of bactericidal compounds plate dilutions of 10-1 to 10-4. Laboratory 2 Bacteriological Techniques Biology 422L -22) Gram stain. Two of the most commonly used characteristics to describe a species of bacterium are its shape and its Gram staining properties. The Gram stain depends on the presence or absence of an outer membrane, extracellular materials, and cell wall composition. Positive and negative Gram stain reactions correlate with differences in cell structure, composition, and permeability and separate bacteria into groups differing in their physiology. Gram staining properties of bacteria are the most uniform with growing cells. In stationary cultures some ordinarily gram positive bacteria may appear to be gram negative. You will be provided with growing cultures of Escherichia coli, Bacillus subtilus, and Staphylococcus aureus. With an inoculating loop place a small drop of the bacterial suspension on a clean microscope slide (clean the slide using a Kim wipe and ethanol). After the drop has dried, fix the bacteria by passing the slide quickly through a Bunsen burner flame. Do not put hot slides on the bench top. Then stain using the following procedure: 1. Cover the smear with alkaline crystal violet (2% crystal violet containing 0.8% ammonium oxalate) - leave 30-60 sec. 2. Rinse with tap water using a Pasteur pipette. 3. Cover with Gram's iodine (0.5% iodine in 2% I) for one minute. 4. Rinse with tap water using a Pasteur pipette. 5. Rinse with 95% alcohol until no more purple dye comes off. Gram positive cells retain the crystal violet. Gram negative cells do not. 6. Rinse with tap water using a Pasteur pipette. 7. Stain with safranin, neutral red, or Bismark brown for 30 seconds to color the Gram negative cells. 8. Rinse with tap water using a Pasteur pipette. Drain and examine using the high power microscope lens. Record the appearance of the cells of each species provided. If you have difficulty focusing on the surface of the slide make a mark with a black marking pen and focus on that first. 3) Streaking of a mixed culture. (Do this immediately after you receive the culture from your TA). During the isolation of a pure culture of an organism from nature and during the maintenance of a culture it is frequently necessary to separate the individual cells from a mixed population. You will be given a culture of a mixture of species. Separate the cells by streaking a drop of the suspension on one or two Laboratory 2 Bacteriological Techniques -3nutrient agar plates. Grow the cells overnight at 37o. The next day you should be able to see colonies differing in appearance; each colony represents the progeny of a single individual in the original mixed suspension. (You can also plate your mixture on McConkey’s agar which is semi-selective for E. coli and on which E. coli has a distinctive appearance). Pick a single colony of each type that is well separated from its neighbors and streak it on a fresh nutrient agar plate. Grow these plates overnight at 37o and check the new colonies to see if they are similar in appearance. You can also use a Gram stain as an additional check that you have isolated a pure culture. 4) Washing side-arm flasks. When you are done with your growth curve you will need to wash your sidearm flasks. These flasks can not be washed in the dishwasher. There is a tendency for materials to be retained in the sidearm and then to poison the culture the next time you use the flask. Kill the culture in the flask with ethanol. Do NOT use disinfectant. It doesn’t wash out. Then rinse the flask tilting it so as to rinse the arm with hot tap water five times, followed by five rinses with distilled water. Hang the flasks on the peg board to dry. Report: 1) Record your observations and data for the growth curve, Gram stain, and streaking of a mixed culture. Plot the growth curve data on semi-logarithmic paper. (Graph paper is included in this manual). Determine the doubling time of the growing cells and draw any other conclusions that you feel are warranted from your data for these experiments (for example, does the composition of the medium influence the growth rate? If so, how? Did the additions made to the cultures kill (bactericidal) or inhibit growth (bacteriostatic) or have no effect on the bacteria?) Make sure to note any cases in which a procedure different from that suggested in the laboratory directions was followed. For this laboratory exercise and the succeeding ones such a brief write-up will be due the week following the laboratory class. 2) Hand in a plate of a pure culture of each of the organisms contained in the mixed culture. BIOLOGY 422L Directions for Using the Microscopes 1. Use a clean glass microscope slide. Label the slide in one corner with the name of the sample to be examined. 2. For a wet mount of live bacteria, place a small (less than 0.05 ml) drop of a liquid culture on the center of the slide. Cover it with a glass cover slip. 3. Using a black marking pen, make a mark on the slide near your sample. 4. Place the slide on the stage of the microscope and hold it in place with clamps if available. Turn on the microscope light. 5. Using the X10 objective locate the black line you made on the slide and focus on it. Then move the slide over to the sample and examine it. 6. After examining the sample with the X10 objective switch to the X40 objective and examine the sample. The jiggling motion of the bacteria is due to Brownian motion. Bacterial motility is faster and more directed. For aerobic bacteria, motility is often dependent on 02 and is usually best observed near the edge of the cover slip. 7. For a microscope with the possibility of moving the condenser and centering the diaphragm use a slide with a marking pen mark on it. Using the X10 objective first focus on the mark on the slide. Close the diaphragm so that its edges are visible. Using the centering screws center the diaphragm. Then adjust the condenser so that the edge of the diaphragm is in focus. Adjust the diaphragm so that it is only open to a diameter just slightly larger than the field of view with the objective in use. On a good microscope with par focal lenses the adjustment for one lens should be good for all objectives. However, on many microscopes you will have to make this adjustment whenever you switch from one objective to another. Name_____________________________ Pre-Laboratory Exercise Laboratory 3 1. If E. coli is given 2 sugars to grow on such as glucose and lactose will it: a. use both at once b. use the glucose first and then the lactose c. use the lactose first and then the glucose d. none of the above. 2. The enzyme ß-galactosidase can carry out the reaction ONP-galactose -----> ONP + galactose. ONP is yellow. In the reactions you will do ONPG is present in large excess. If 0.1 ml of a bacterial extract has enough enzyme to produce enough ONP to give an OD of 0.2 after an enzyme assay time of 10 min, then a. What will the OD be after an enzyme assay time of 20 min? ______ b. What will the OD be after 10 min if I add twice as much extract? ______ c. What will the OD be after 10 min if I add 3 times as much ONPG? _____ d. What will the OD be if I take the extract from cells grown with glucose rather than lactose? ___________. 3. What would the difference be between measuring units* of enzyme/OD of cells and total units of enzyme/ml with a culture growing on lactose alone? What will happen to each measurement if the number of cells doubles but the amount of enzyme per cell is constant? *(Enzyme activity is expressed in arbitrary units. For example, one Miller unit of βgalactosidase activity is that amount of enzyme which will result in the production of 0.001 OD420 of ONP from ONPG per minute.) Note. This lab takes a long time. Biology 422L Laboratory 3 Enzyme Induction Objectives: 1) To learn how to perform enzyme assays. 2) To observe diauxic growth. 3) To observe enzyme induction. Reference: Slonczewski and Foster 345-351. The regulation of the enzymes involved in the metabolism of lactose is the most thoroughly studied and one of the best understood control mechanisms in bacteria. The genes for three proteins are involved: β-galactosidase (z), lactose permease (y), and beta-galactoside transacetylase (a). These genes form an operon; that is, their activity is controlled by an operator locus (o) adjacent to them. There is a promoter site (where RNA polymerase binds) just to the left of the operator. The regulator gene (R) which codes for the synthesis of the repressor for this operon is located next to the operator gene. It has its own promoter site (P'). P' R | P |o | z | y | a regulator gene lac operon Map of the lac operon When E. coli is grown on glucose the lac operon is ordinarily repressed and the cells contain little β-galactosidase. When the cells are transferred to lactose the operon becomes depressed; βgalactosidase, permease, and transacetylase are synthesized; and the cells grow. 1) Growth of the cultures. 1. Put 5 ml of medium A into a sterile side arm flask. 2. Add glucose to 0.005%, 0.25 ml of 0.1% glucose solution in 5 ml of medium A is 0.005% glucose. 3. Add lactose to 0.2%. For example, add 0.5 ml of a 2% lactose solution to 5 ml of medium A+ glucose to arrive at a final concentration of 0.2% lactose. 4. Add 0.1 ml of a late log-phase E. coli culture in LB. 5. Read the OD600. Be sure to first "zero" the spectrophotometer with a tube containing only the growth medium. BE SURE TO WIPE OFF FINGERPRINTS! NOTE THE TIME AS TIME ZERO. 6. Place the flask in the 37o shaking water bath. Laboratory 3 Enzyme Induction -27. Observe and record OD600 of the culture every 15-20 min. Record the time and the OD. 8. After 15-20 minutes have elapsed, withdraw a 0.5 ml sample of culture after reading the OD600. Process this sample as soon as possible as described below under "β-galactosidase assay". Put the flask back on the shaker and read the OD every 15 min and collect a 0.5 ml sample every 30 minutes after reading the OD600. Be sure to label the tubes with the sampling time. 2) β-galactosidase assay. 11. Put 0.5 ml of collected culture in 2 ml of Z buffer. 12. Add 4 (do not add more) drops of CHCl3 (chloroform) and 2 drops of 0.1% SDS (sodium dodecyl sulfate also called sodium lauryl sulfate)). Do this on the table by the window fan. 13. Vortex tube vigorously for 1 min. and place on ice, if available, until all samples are collected. REMEMBER TO INCLUDE A "BLANK" CONTAINING MEDIUM ONLY. Then take the tubes to the 37o roller drum and evaporate off the chloroform (do not put a lid on the tube). This will take about 15 min. Check to see that no drops of chloroform are visible at the bottom of the tube. 14. Place samples in 30o water bath, add 0.4 ml of 4 mg/ml ONPG (orthonitrophenyl βgalactopyranoside). Note the time at which you add ONPG. 15. Note that there are 2 times which should be recorded for each sample. The time the sample was taken and the number of minutes the enzyme assay was run. Be careful to record both of these times and to distinguish between them in making calculations. 16. Watch for yellow color development, which may appear quickly or slowly, depending on the amount of β-galactosidase present. When the tubes are obviously yellow, terminate the reaction by adding 0.5 ml of 1 M Na2CO3 solution. Mix. If the tubes do not become yellow, let reaction continue for 30 min, then add Na2CO3 (this raises the pH). NOTE THE TIME ELAPSED BETWEEN THE ADDITION OF ONPG AND ADDITION OF Na2CO3. 17. Read OD550 and OD420 for each tube. Don't forget to "re-zero" the spec between wavelength changes. Determine the relative amount of enzyme accessible to ONPG by measuring the amount of ONP (orthonitrophenol) produced. The amount of ONP is a linear function of OD at 420 mμ for OD's up to about 1.5. Make a correction for OD due to light scattering rather than to ONP by also reading the OD at 550 (scattering increases with decreasing wavelength so the corrected OD is OD420-1.75 [OD550]). Once the reaction has been stopped with Na2CO3 you may wait as long as 30 min to read the OD's of your tubes. Laboratory 3 Enzyme Induction -3ONPG β-galactosidase → ONP + gal The amount of orthonitrophenol produced in the reaction is a function of the amount of enzyme available to catalyze the reaction. Twice as much enzyme will convert twice as much ONPG to ONP in the same amount of time (provided there is plenty of ONPG available). The amount of ONP produced is also a function of the length of time which the ONPG is allowed to react with the enzyme. Again, generally in twice as long an incubation with ONPG the enzyme will produce twice as much ONP. Calculate the units of enzyme per OD of cells (at that time) by using the following formula: Units of enzyme ------------------------OD600 of cells at time t = 1000 [OD420 - 1.75 (OD550)] -----------------------------------------------------------(min of enzyme assay) X (OD600 of cells at time t) Report: Give a table showing your data. Plot the growth curve on semi-log paper. Plot units of enzyme/OD of cells vs. time on regular linear graph paper. Explain your results (i.e., when was βgalactosidase present in large amounts and when was it absent, and why? How does this reflect how the cell regulates expression of the lacZ gene?) Notes: (Numbers refer to numbers in the directions above). 3) Cell growth. 4. It is important to use very vigorously growing cells, or else the lag time before growth resumes may be excessive. 5. Fingerprints will increase the apparent OD600 of the culture. One must always "zero" the spec with growth medium only to establish a baseline. 6. Rapid cell growth is needed here, which for E. coli means 37o and good aeration. 7 & 8. Do not allow the cultures to sit around at room temperature, they won't like it. 4) β-galactosidase assay. 12. CHCl3 and SDS (a detergent) will cause the cells to lyse, liberating the β-galactosidase. Z buffer is a pH 7.0 phosphate buffer. Laboratory 3 Enzyme Induction -414. ONPG is a compound which can be cleaved by β-galactosidase, resulting in a yellow product (orthonitrophenol) which can be quantified spectrophotometrically. 15. Substrate for β-galactosidase (ONPG) was added in huge excess in step 4, so how fast color develops depends on how much β-galactosidase is present. Addition of Na2CO3 raises the pH sufficiently to cripple the β-galactosidase. 16. The OD420 which you measure needs to be corrected for light scatter due to the debris from the broken cells; to do this you measured the OD550 ( a wavelength at which ONP doesn’t absorb light) and multiplied this OD by 1.75 to determine the light scatter at 450 nM. (Note that light scatter increases with decreasing wave length of light; see your physics book). WARNING THE PRELAB FOR THE NEXT WEEK REQUIRES TIME AND THE USE OF THE TEXT. WARNING THIS PRELAB EXERCISE REQUIRES TIME AND WORK WITH THE TEXT. Name_________________________ Enrichment Cultures - Preliminary to Laboratory 4 Match the bacterial type with the inoculum (source of the bacteria) (column I) and the medium (II) and growth conditions (III) you would use to enrich for that bacterial type example (l.s.A.). Use the directions for lab #5. Try to do as much as you can without the book. Then complete the exercise using your text. You must hand in the answers to this exercise before you begin the experiment. This exercise is designed to help you understand enrichment cultures and the wide variety of situations in which they can be used. We will actually do only a few of these examples. I Bacterial Type 1. Rhizobium 2. __s__ II Inoculum __A__ q. Yogurt Lactic Acid Bacteria r. Wine 3. Propionic Acid Bacteria s. Root nodules 4. Pseudomonas w. Air 5, Azotobacter x. Swiss cheese 6. Nitrosomonas y. Boiled soil 7. Acetobacter z. Soil or mud (use more than once) 8. Thiobacillus 9. Thiobacillus denitrificans 10. Non-sulfur Photosynthetic Bacteria 11. Sulfur Photosynthetic Bacteria 12. Bacillus cereus & other aerobic spore formers Enrichment Cultures Preliminary to Laboratory 4 Biology 422L -2III Medium C source N source and Growth Conditions Other Components Aerobic Light --- yes no A Nutrient agar* B Glucose Yeast Extract --- no no C Lactate Yeast Extract --- no no D Malate NH4Cl --- no yes E NaHCO3 NH4Cl Na2S no yes F Ethanol Yeast Extract --- yes no G --- NH4Cl Na2S2O3 yes no H NaHCO3 NH4NO3 Na2S2O3 no no I Mannitol --- --- yes no J CaCO3 NH4Cl --- no no K Benzoic acid NH4Cl --- yes no *Nutrient agar will be used for those cases in which the primary enrichment is the choice of inoculum. Biology 422L Laboratory 4 Enrichment Cultures Objectives: 1) To learn how to isolate various types of bacteria from nature. 2) To become familiar with some of the many different types of bacteria. Reference: Slonczewski and Foster, chapters 14, 18, 21, and 22. The Prokaryotes . Bergey’s Manual of Determinative Bacteriology. Report: Describe the bacteria you obtained from each isolation. Include gram stain, colony morphology, growth rate, bacterial shape, size, motility, and any other distinguishing features. The report for this laboratory will be handed in as two parts. The first part containing propionic acid bacteria and spore formers will be due in 2-3 weeks. The second part covering the remaining bacteria will be due in 4-5 weeks. The technique of enrichment cultures is based on providing a specialized environment for bacterial growth, and using an inoculum which contains many kinds of bacteria (including the one desired). The bacteria which can grow best under the conditions provided will multiply fastest and become the most numerous. If a small bit of the culture is then placed in fresh medium the bestadapted bacteria will continue to grow fastest (this is a case of true Darwinian survival of the fittest). Thus the bacteria which are present at the end of several successive enrichments may represent only a very small portion of those species initially present. To take an extreme example, if the medium contains no nitrogen source, then only nitrogen-fixing bacteria (which can use N2 from the air) will be able to grow. You may bring soil or mud samples with you to the laboratory. Record the type of environment you took them from. Each student should try all the enrichment procedures since some will be unsuccessful with any particular soil sample. For aerobic conditions, put the inoculum (a small pinch) in a small amount of liquid medium (less than 1/2 inch) in the bottom of a sterile tube or vial or streak the inoculum on an agar petri plate. For anaerobic conditions put the inoculum in a sterile plastic tube and add about 3 ml liquid medium. Use paraffin to seal the culture from the air. To enrich a second time break the seal with a spatula and transfer a small drop with an inoculating loop or Pastuer pipette from the culture to a second identical set-up containing fresh medium. There will be a table on the board indicating the average progression of the cultures. Consult it to be sure you don’t forget anything. After enrichment streak all aerobic cultures on plates and note colony form and color. Make gram stains of all enrichments and record shape, size, and staining properties of the cells. Also look at wet mounts and at wet mounts with a drop of India ink (to visualize capsules) and record presence or absence of a capsule, and any other distinguishing features. Test for the presence of catalase by adding a drop of H2O2 to a colony and looking for the formation of O2 (bubbles). Keep a record of the procedures followed for each enrichment. This experiment will take weeks to complete. Laboratory 4 Enrichment Cultures I. 2 Propionic Acid Bacteria Swiss cheese (use the less aerobic middle) Liquid A medium with 2% sodium lactate and 1% yeast extract, pH 7.0. A medium contains K2HPO4 - 10.5 g/l, KH2PO4 - 4.5 g/l, (NH4)2SO4 1 g/l, sodium citrate.2H2O - 0.5 g/l and 10 ml of 20% MgSO4.7H2O added after autoclaving. Conditions: Anaerobic, room temperature, dark To obtain an anaerobic environment place 3 ml of medium in a white plastic tube, add your inoculum (this should be small) and seal the top with paraffin (no air bubbles). Check in two days. Enrich twice in liquid medium using anaerobic growth conditions. These bacteria convert lactate to propionic acid. Look up propionic acid bacteria in your text and compare your isolates with those described in the text. II. Organism: Inoculum: Medium: Organism: Inoculum: Medium: Non-sulfur photosynthetic Bacteria Soil or mud Van Niel's. MgSO4.7H2O - 0.2 g/l, K2HPO4 - 1.0 g/l, FeSO4.7H2O - 0.01 g/l, CaCl2 - 0.02 g/l, MnCl2.4H2O-0.002 g/l, Na2Mo04.2H2O- 0.001 g/l, NaCl- 0.5 g/l, sodium malate-5.0 g/l, yeast extract - 0.5 g/l, NH4Cl - 1.0 g/l. pH 7.0 - 7.5 Conditions: Room temperature, anaerobic, about 2 feet from a light source such as a 60 watt bulb. To obtain an anaerobic environment place 3 ml of medium in a clear plastic tube, add your inoculum and seal the top with paraffin (no air bubbles). Check in two weeks. Enrich using the pink region of your culture. These bacteria grow slowly. Non-sulfur photosynthetics should be pink, red, or purple. Malate acts as a carbon source and as an electron donor. Look up these bacteria in your text and compare your isolates with those described. Note color, size, gram-stain, and any other distinctive characteristics. III. Organism: Inoculum: Medium: Conditions: Bacillus cereus and other aerobic, spore-forming bacteria. Place one cc of soil, a pinch of soluble starch and 3 ml of 1% yeast extract in a long glass tube. Place the tube in a boiling water bath for 3 min. Nutrient agar plates Room temperature, dark, aerobic Streak about 0.1 ml of the boiled inoculum on nutrient agar plates. After 1-2 days examine cells from colonies using wet mounts to look for spores and for motility, and using gram stains. The distinctive rhizoid growth of B. cereus should allow you to pick it out from other species. Only spore formers can survive boiling. Compare your isolates with those described in the text. Laboratory 4 Enrichment Cultures IV. Organism: Inoculum: Medium: Conditions: 3 Pseudomonas A very small pinch of soil Liquid. Mg2SO4. 7H2O - 0.2 g/l, K2HPO4 - 1.0 g/l, FeSO4.7H2O 0.05 g/l, CaCl2 - 0.02 g/l, MnCl2.4H2O - 0.002 g/l, Na2Mo04.2H2O 0.001 g/l, benzoic acid - 4.0 g/l, NH4Cl - 1.0 g/l, pH 7.0 Aerobic, dark, room temperature; 1-2 ml of medium in a short screw cap tube. In 2-3 weeks enrich into a second tube of 1-2 ml of medium. Very few species of bacteria can grow on benzoic acid as a carbon source. Note that it is not possible to give you precise directions for this lab as the exact of species of bacteria isolated will vary from year to year. Thus the growth rate and other properties of the bacteria will have to be determined by you. You will need to keep careful track of your cultures and see how they grow this year. type of bacteria source Working Record colony color growth rate and characterization microscopic observations morphology liquid solid aerobic/ bacterial shape motility anaerobic and size propionic acid bacteria Non-sulfur photosynthetic bacteria N/A spore forming bacteria N/A N/A Pseudomonas N/A N/A Gram stain capsule other characteri stics type of bacteria source colony morphology color Report to Hand In growth rate and characterization microscopic observations liquid solid aerobic/ bacterial shape motility anaerobic and size propionic acid bacteria Non-sulfur photosynthetic bacteria N/A spore forming bacteria N/A N/A Pseudomonas N/A N/A Gram stain capsule other characteri stics Laboratory 4 Enrichment Cultures -5Question for part 1. Can you identify any of the genera and/or species of spore formers you isolated using your book, The Prokaryotes, and Bergey's Manual? Question for part 2. Explain how the enrichment culture for the isolation of nonsulfur photosynthetic bacteria works. Why did most people observe faster growth of these bacteria in the subculture than in the original culture? Name_______________________ Pre-Laboratory Exercise Experiment 5 Which do you expect to be more common cellulose-over producing or cellulose-minus mutants? Why? If Agrobacterium contains 4,000 genes and you obtain a frequency of 0.2 % for cellulose-minus mutants how many genes do you think might be required to synthesize cellulose? (Remember that mutants which are unable to grow on minimal medium will not be recovered). Biology 422L Laboratory 5 Transposon Mutagenesis Objectives: 1) To learn how to introduce a transposon into a bacterium, 2) To learn how to screen for a desired bacterial mutant. Reference: Slonczewski and Foster, Chapter 9. Transposons can be introduced into bacteria via phages or plasmids. Wherever a transposon integrates into the bacterial DNA it causes a mutation. Mutations due to the insertion of a transposon have the advantage that they are usually easy to find and clone since the transposon is a relatively large piece of DNA. The transposon we will use in this experiment is miniTn5 which is about 4kb in size. It contains inverted repeats on the ends and encodes an enzyme which makes the cell resistant to tetracycline. In addition it has a promoterless green fluorescent protein (gfp) coding sequence with a ribosome binding site at one end. Thus if the transposon integrates behind a promoter the gfp gene will be expressed under the control of that promoter. This transposon has no specificity in its integration site. We will introduce the transposon into Agrobacterium tumefaciens LTU50 using a plasmid which is conjugative and carries the transposon, and carbR and the transposase both located outside the transposon. This plasmid has an origin of replication which is recognized by Escherichia coli but not by A. tumefaciens. The E. coli strain is a multiple auxotroph and will not grow on minimal medium. The A. tumefaciens strain will grow on minimal medium and is sensitive to tetracycline and resistant to chloramphenicol. We will conjugate the plasmid from E. coli into A. tumefaciens. We will then select for A. tumefaciens which have received the transposon by plating the mating mixture on minimal medium containing tetracycline. Neither parent strain will grow on this medium. We will then screen the A. tumefaciens containing Tn5 insertions in which gfp is expressed and for mutations in the ability to synthesize cellulose by growing the bacteria on medium containing the dye cellufluor which stains cellulose to give a bluewhite fluorescent color under UV light (this is the dye in your white cotton shirt or lab coat which also causes them to fluoresce under UV light). Colonies which are darker (or brighter) than the average are most probably mutants which fail to produce (or overproduce) cellulose. Gfp will fluoresce a pale yellow green under UV. In addition we will screen for mutants which are altered in their ability to produce curdlan (poly-β-1,3-D-glucose) using plates containing the dye aniline blue which turns navy blue in the presence of curdlan. Note that plates containing aniline blue will show little under UV light as aniline blue interferes with the UV detection of Gfp and cellufluor fluorescence. This technique of obtaining bacterial mutants by screening for the desired characteristic of the mutant cells has been much used particularly in obtaining mutants in virulence factors of human and plant pathogens. FIRST WEEK FIRST DAY 1. You will be given a culture of the E. coli strain S17 pUTminiTn5gfp (carbR, tetR). The plasmid carries Tn5. You will also be given a culture of A. tumefaciens strain LTU50 cmR. Take the S17 culture and add 0.25 ml to 5 ml of Luria broth containing carbenicillin (50 μg/μl) in a side arm flask. Read the OD550 and grow the bacteria on a shaker until the OD550 doubles. This will give you a log culture of the bacteria which is required for efficient conjugation. Then place about 0.5 ml of each culture on the surface of a Luria agar plate. Tilt the plate and rotate it gently to mix the cultures. Incubate the plate overnight at room temperature right side up. Laboratory 5 Transposon Mutagenesis -2FIRST WEEK SECOND DAY 1. Add 2 ml of phosphate buffered saline to your conjugation plate. Sterilize a glass rod in ethanol and scrape the cells from the surface of the agar and make a suspension. Pipette the suspended cells into a sterile Eppendorf tube. 2. Spin the tube in the centrifuge for 1 min. to pellet the cells. Pour off the supernatant into a container of disinfectant. 3. Add 1 ml of phosphate buffered saline to the tube and resuspend the cells by vortexing them. 4. Plate 0.1 ml of the cells undiluted and of a 10-1 dilution in PBS onto Luria agar plates containing chloramphenicol (20 μg/ml) and tetracycline (10 μg/ml). Incubate the plates at room temperature. Also plate 0.1 ml undiluted onto a buffered yeast extract with 2% glucose plate containing chloramphenicol, tetracycline, and aniline blue. Phosphate buffered saline (PBS) Na2HPO4 (anhydrous) HK2PO4 NaCl MgSO47H2O H2O dissolve each salt in order 7g 3g 4g 0.2g to 1L SECOND WEEK FIRST DAY 1.Observe your plates. Mark any colonies with unusual colony morphology as these may be altered in exopolysaccharide production. Cellulose and curdlan are both exopolysaccharides. Check you aniline blue plates using visible light for colonies which differ in color from the majority of colonies (do not check these plates with UV light as aniline blue prevents the observation of UV fluorescence). Use sterile toothpicks to pick colonies that you marked onto 2 plates of minimal medium containing chloramphenicol, one with and one without cellufluor (0.001%) and onto a fresh aniline blue plate. A grid pattern to use in picking your colonies is given in the manual. If more than one size or morphology of colony is apparent be sure to pick some of each different type. If control cultures are available, in the bottom corner of your plates place an inoculum of a cellulose-plus, chloramphenicol-resistant and a cellulose-minus, chloramphenicol-resistant A. tumefaciens strain as a control. Carefully mark the location of the control inocula. Incubate the plates at room temperature. SECOND WEEK SECOND OR THIRD DAY 1. Observe and record the growth of colonies on your plate. Use reflected UV light (be careful to wear a face shield)to observe your Luria agar plates and mark any isolates which appear yellow green due to expression of Gfp. (These will be rare and only a few people will have any). Also Record how many are cellufluor bright or dark and how many express detectable gfp. 2. Record how many colonies are altered in curdlan production. Curdlan will turn colonies on an aniline blue plate navy blue. The parent strain used in this experiment is used commercially for curdlan production and is a curdlan over-producer. 3. Report your data and the class data. Calculate the frequency of obtaining bright and dark mutants, aniline blue staining mutants, and of obtaining insertions in which Gfp is expressed at a detectable level from your data and from the pooled data of the class. Hand in a one to two page report stating how the protocol works to give Tn5 mutants altered in cellulose synthesis, curdlan synthesis, and how this promoter probe transposon lets you pick insertions in genes which are expressed under the growth conditions you used . Describe your results and hand in the plate of the mutants which you isolated. Questions to be answered. Tn5 has no sites for EcoRI. How could you clone the DNA containing the Tn5 insertion from your mutants? Write out a protocol for this cloning using vectors described in your book or in lecture. Indicate the media to be used to grow the bacteria. You do not need to give details of methods for such procedures as DNA extraction or bacterial transformation. Simply state "Extract the DNA from....". Name______________________ Pre-Laboratory Exercise Laboratory 6 Suppose that substance IGLOO is required for the formation of the bacterial cell wall. It is made by the following pathway: A B C D Ig + L Igl + u Iglu Iglou Igloo A bacterium which is unable to make enzyme A will be unable to grow. Will it grow if I feed it Igl? _______ Will a bacterium which can't make enzyme C grow if I feed it Igl? ________ The A bacterium accumulates Ig. Will it be able to cross feed C? _______ The C bacterium accumulates Iglu. Will it be able to crossfeed A? _____ Biology 422L Laboratory 6 Biochemical Pathways Objectives: 1) To become familiar with the growth characteristics and interactions of mutant bacteria blocked in a particular biochemical pathway. 2) To experience how bacterial mutants can be used to help to elucidate a particular pathway. 3) To become familiar with the concept of a biochemical pathway and to understand the implications of this concept. References: any biochemistry textbook, and the biocyc web site http://biocyc.org/server.html Look up E. coli K12 and arginine synthesis. Auxotrophic mutants have been used to help elucidate the pathway for the biosynthesis of many compounds. You will be provided with a set of four auxotrophic mutants which require arginine (an amino acid) for growth. These mutants are point mutants. When they were tested in pairs in a cis/ trans complementation test they were each found to complement the other three mutants. Thus each of these mutations affects a different cistron or polypeptide chain (remember that some enzymes are composed of multiple subunits or polypeptide chains). We will use two different techniques to help us to determine the biosynthetic pathway for arginine in E. coli and the rough location in that pathway of the enzyme encoded by each of these genes. Growth on suspected intermediates in arginine synthesis. Arginine is made from glutamate. Ornithine is known to be involved early in the pathway leading to arginine and citrulline is a possible later intermediate. You will be given a plate with the four Arg mutants (H, Y, P, and C). One person at each bench should do this part of the experiment and incubate it where everyone can read the results. Using a bacteriology loop take a small amount of bacteria of each strain from the plate and resuspend them in buffer. Use the bacteria suspended in buffer to streak plates containing glucose minimal medium, glucose minimal medium plus ornithine, glucose minimal medium plus citrulline, and glucose minimal medium plus arginine (4 plates for each bacterial mutant). Observe the growth of the mutants the next day. Growth on a particular substrate is proof that the bacteria can make arginine from that substrate and so must be defective in some earlier biochemical reaction. Lack of growth on a substrate may only indicate a failure of the cells to be permeable to that substrate. (Note that these mutants are point mutants and can revert; thus the small number of colonies you will see on some plates probably represent revertants of the mutation). Cross-feeding. (Each student should do this part) Many biosynthetically defective cells accumulate the substance which they can no longer metabolize. If this happens then another mutant defective in an earlier biosynthetic reaction may be able to grow on the intermediate accumulated by the first cells (provided the first cells release the intermediate into the medium and that the second mutant is permeable to the intermediate). This is the basis of cross-feeding experiments. Laboratory 6 Biochemical Pathways -2Each student should do this part of the experiment. Streak the four arginine-requiring mutants on glucose minimal medium so that each mutant is near each of the other three but be careful that the mutants do not touch each other. Use bacteria from the plates and make very heavy streaks on the surface of the agar. A possible streaking pattern is illustrated on the bottom of this page. Streak the cultures very close together. Incubate the plates at 37o. If you made your streaks close together record the data the next morning. If you made light streaks far apart record the data the next afternoon. Check your plates again the next day. Note that substances released by one mutant will form a gradient and thus cross-feed those cells nearest it first. Also note that if you leave your plates too long the intermediates will diffuse all over the plates and everything will grow. Report: Record your data in a table. Using the data from the tests you have made, attempt to order the mutants in the arg pathway. Indicate which mutants could cross-feed which other mutants. Also attempt to order the mutants with respect to ornithine and citrulline. Now look up the pathway for arginine biosynthesis in a biochemistry book (for example, Henry R. Mahler and Eugene H. Cordes. Biological Chemistry. 2nd edition, 1971. pp. 776-777 call number QP514.2 .M35 1971 or you can find the pathway at the BioCyc web site http://biocyc.org/ Name__________________________ Pre-Laboratory Exercise Laboratory 7 A solution containing 109 phage particles per ml was mixed with 109 E. coli. The bacteria were removed from the solution by centrifugation after 3 min and the number of phage particles in the supernatant was found to be 1 x 108 per ml. What happened to the other 9 x 108 phage per ml? The bacteria were incubated with the phage for 25 min and then removed by centrifugation. The number of phage particles in the supernatant was found to be 9 x 1010. What was the burst size of the phage (i.e. how many phage progeny did the average phage produce)? Biology 422L Laboratory 7 Phage One-Step Growth Objectives: 1) To learn how to do a plaque assay to count the number of viable virus particles. 2) To observe the growth of a lytic bacteriophage. References: Slonczewski and Foster Chapters 6 and 11. The study of the lytic double-stranded DNA phages T1-T7 played a major role in the development of molecular biology. These viruses have simple genomes of 25-150 genes. When the virus infects a cell the protein coat remains outside the cell and only the DNA enters the cell where it is transcribed and translated into proteins. The details of the life cycle and genetics of the T phages are described in your text. Most viruses are too small to be seen in the light microscope. The total number of virus particles in a preparation can be determined by visually observing and counting the virus particles in the electron microscope. The number of viable virus in a preparation of a lytic virus can be determined by a plaque assay. This assay is the equivalent of the viable cell count used to determine the number of live bacteria. In this experiment we will determine the number of live T7 virus in a solution. The virus will be added to a growing culture of E. coli in a ratio of approximately 1:1 and the culture observed until it lyses. Then the number of T7 virus will again be determined. The comparison of the initial and final number of T7 will allow you to determine the average number of T7 produced by the infection of a single bacterium. The number of viable phage particles in a suspension can not be determined by plating the phage directly on agar as is done with bacteria. Instead the phage are mixed with a large number of bacteria (so that most bacteria have no phage on them) to which they adhere. The bacteria/ phage mixture is then placed in agar and poured onto a petri plate (the agar immobilizes the bacteria and phage so that they do not move around on the plate). Most of the bacteria plated have no adherent phage and simply divide to produce a confluent layer or lawn of bacteria covering the plate. In those bacteria with absorbed phage particles the phage grows and lyses the bacterium releasing many new phage which absorb to neighboring bacteria, grow, and lyse these bacteria. This process produces a hole in the bacterial lawn at each location where one phage was originally present. With T7 phages these holes or plaques continue to grow in size with continued incubation so the number of plaques must be scored soon after they become visible. Eventually T7 will lyse all of the bacteria on the plate. Laboratory 7 Phage One-Step Growth -2NOTE all plating in this experiment is done to determine number of phage and should use soft agar plating techniques. 1) You will be provided with a fresh culture of E. coli 011Y thy-. Place 5 ml of growth medium containing 10 g/l tryptone, 5 g/l NaCl and 40 mg/l thymine into a sterile side-arm flask. Add E. coli to obtain an OD550 of about 0.2. Grow the cultures in the 37o water bath with shaking until the OD reaches 0.3 to 0.4 (or almost doubles from the initial OD). This will result in a log phase culture of the bacteria which is required for the efficient growth of the phage. Read and record the OD's after various times. 2) While the bacteria are growing prepare 21 sterile tubes containing 0.9 ml of 0.5 M NaCl. 3) When the bacteria have roughly doubled in optical density add 0.1 ml of the stock solution of T7 with which you will be provided to the flask. Mix the stock before you add it. Mix the flask and allow it to sit on the bench top for about 1 min. Then remove about 0.7 ml from the flask and place it in an Eppendorf tube. Read the OD of the flask and record it and the time and place the flask back on the 37o shaker. 4) Take 0.1 ml from the Eppendorf tube and add it to 0.9 ml of NaCl in one of the tubes you made. This is a 10-1 dilution of the total T7 present initially. 5) Centrifuge the rest of the contents of the Eppendorf tube for 3 min to pellet the bacteria. (Adsorbed phage pellet with the bacteria, while free phage remain in the supernatant. These free phage will not contribute to the growth of the phage.) 6) Dilute the total T7-bacteria mixture (#4) in steps of 10 to 10-6. You will wish to plate out the 10-4, 10-5, and 10-6 dilutions. This will allow you to measure the initial total number of phage. 7) Procedure to titer phage (soft-agar plating). You will be given an early stationary phase culture of E. coli 011 sitting at room temperature. Put a sterile 25 ml pipette in this culture. You can leave the top off and use the same pipette for all your work. Now take 1 sterile white capped tube. Add 0.1 ml of the appropriate phage dilution; then add 0.25 to 0.5 ml of supplied E. coli 011 stationary phase culture (the exact amount does not matter). Then add 3-4 ml of soft agar from the bottle in the 55o water bath. Mix quickly by rolling the test tube in your hands and pour onto your plate. Tilt the plate immediately to cover the surface with the soft agar. Then allow it to stand upright to solidify. Your laboratory instructor will demonstrate these techniques. 8) Dilute the supernatant from centrifuging the bacteria-T7 mixture in steps of 10 to 10-6. Plate 10-4, 10-5, and 10-6 dilutions using soft agar. This will determine the number of free unabsorbed virus remaining. Note the plating in steps 6 -8 must be done within 10 minutes of adding T7 to the culture. To determine the -3- number of phage which adsorbed to the bacteria simply subtract the number of free phage from the total (measured in #6). 9) Read and record the OD of your culture at various times after adding the phage. When the OD decreases (or if the OD does not decrease after 45 min of incubation with T7 add 1 drop of chloroform to lyse the cells) remove your culture from the shaker and determine the number of T7 present by diluting the culture in 0.5 M NaCl in steps of 10 to 10-8. Plate 10 -5, 10-6, 10-7, and 10-8 dilutions using soft agar. This will determine the number of T7 present after one cycle of growth. 10) When your plates have solidified, turn them over and incubate them overnight at room temperature in your drawer (or for faster growth at 370; plates must be scored in 20 hours or less if this temperature is used). 11) Count the number of plaques on your plates the next morning. Report: 1) Report your data in a Table. 2) Plot the growth curve of the bacteria on semi-log paper. Indicate the time of addition of T7 and the growth phases of the virus. 3) Determine the percent of T7 absorbed to the bacteria after 1 min incubation. Determine the average burst size for T7, how many virus particles does the average infected cell release? Don't forget to count only phage that absorbed to the bacteria in calculating the initial number of phage which grew to yield the final number. Burst size = Final number of phage Initial absorbed phage = Final number of phage Initial total phage - unabsorbed phage See the description in your text page. 4) Given a mammalian cell line which grows as a monolayer in culture dishes, how could you modify this assay to determine the number of viable particles of a lytic mammalian virus? Of an oncogenic mammalian virus? Name ______________________________ Pre-Laboratory Exercise Laboratory 8 part 1 1. Go to the web site www.biocyc.org and ask a TA or Dr. Matthysse to show you how to use this web site. 2. Using the tools found at www.biocyc.org look up the genes for tryptophan biosynthesis in a K12 strain of E. coli and in Agrobacterium tumefaciens strain C58. The expression of these genes in E. coli is regulated by a leader peptide sequence. Do they appear to be regulated by a leader peptide sequence in C58? 3. Could I grow E. coli cells on minimal medium with tryptophan if I made an insertion mutation in trpE? trpC? trpB? Why or why not? 4. Could I grow C58 cells on minimal medium with tryptophan if I made an insertion mutation in trpE? trpC? trpB? Why or why not? Name _____________________________ Pre-Laboratory Exercise Laboratory 8 part 2 1. Hand in an outline of the experiment you propose to carry out (on a separate sheet of paper). What mutant phenotype are you trying to find? How will you screen or select for it? What media and solutions will you need to make? Are there other phenotypes which may be associated with your mutant and for which you would like to test the mutant? If so, how will you do this? 2. Can you select for the mutant you want to find? If so, how? If not, how will you screen? How many mutants could you screen using your proposed technique? How frequent do you expect the mutant you want to find to be? Is the experiment feasible? Check the genes and pathways you wish to mutagenize in the Agrobacterium tumefaciens C58 genome using the Biocyc web site. Laboratory 8 Biology 421L Transposon mutants of A. tumefaciens: a real experiment In this experiment we will be doing a real experiment: that is the information which we will obtain is unknown and the results to be expected are not known. The information which we obtain will be useful in assigning functions to genes in the sequenced A. tumefaciens genome. We will use transposon mutagenesis to obtain mutants with desired characteristics in A. tumefaciens. If there were time, we would also clone and sequence the DNA adjacent to the insertion to determine the site of the insertion (Since A. tumefaciens has a sequenced genome, we can determine the gene in which the insertion is located from a short DNA sequence next to the transposon). The general outline of the experiment we will undertake is as follows: 1. Make competent A. tumefaciens and introduce the plasmid pRL27 which carries a transposon into the bacteria. The plasmid carries an origin of replication which is not functional in A. tumefaciens. 2. Select A. tumefaciens which are resistant to kanamycin (carried on the transposon). 3. Select or screen for mutants with the desired characteristics. Your group will need to decide on a type of mutation which you wish to investigate. The phenotype should be easy to determine by selection or screening. Possibilities include (but are not limited to) nonmotile mutants, mutants which produce more or less of a polysaccharide which can be stained by including a dye in the plates, mutants which can or can not grow on certain substrates, and mutants which require a particular substance to grow. Once your group has decided on a type of mutant which you would like to obtain you will need to design the selection or screening protocol and make the appropriate media or reagents. Obviously the type of mutant you choose will be determined by the ease of the method of detecting the mutation. (Note that the lab does have velvets which you can use for replica plating). 4. Obtain a mutant with your desired phenotype and confirm the phenotype. Characterize the mutant with respect to any other interesting phenotypes. 5. If you had time, the next step would be to determine the site of the insertion which gave rise to the mutation. To do this, you would need to isolate DNA from the mutant and digest the DNA with a restriction enzyme which does not cut within the transposon. Because this artificial transposon carries an origin of replication within it and a gene for resistance to kanamycin you could then just ligate the DNA to seal the linear pieces you obtained by the digestion into circles and use the ligated DNA to transform E. coli. Select the clone which carries the plasmid made from your transposon and the adjacent DNA by plating on kanamycin. Purify the plasmid DNA and send it for sequencing using primers directed outward from the ends of the transposon. Since it is unlikely that you will have time to complete this part of the study you should make a stock of your mutant and careful records of everything you did. This information and the mutant you obtained will be used next year in this part of the lab by a new group of students. Alternatively, if you are interested, it may be possible for you to continue your study of this mutant next semester as a research or special topics project. As you would do for any research you were conducting, you should begin by looking up references. The plasmid you will use to make your mutants is described in R. L. Larsen, M. W. Wilson, A. M. Gross and W. W. Metcalf. 2002. Genetic analysis of pigment biosynthesis in Xanthobacter autorophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178: 193-201. You should also look up what is known about the A. tumefaciens genome using biocyc http://biocyc.org/ and entrez on the NCBI web site http://www.ncbi.nlm.nih.gov/ to search for genes in which you are interested. Laboratory 8 Part 1 Design of your experiment and preparation of materials Design of your experiment You will need to determine what type of mutant you wish to try to obtain and characterize. This could be a mutant which is nonmotile, which over- or under-produces a particular polysaccharide, which can grow on a particular substrate, or which can not grow on a particular substrate, or which requires a particular substance for growth or is resistant to some potentially toxic substance or any other characteristic for which you can screen. Examples of mutants and required media are as follows: Mutant Nonmotile Overproduction of curdlan Underproduction of beta-linked polysaccharides Inability to grow on agar Inability to grow on sucrose Requires an amino acid to grow Medium needed motility agar with kan buffered medium with aniline blue and kan medium with cellufluor and kan agar medium with no carbon source and agar medium with sucrose medium with glucose as a carbon source and kan and medium with sucrose as a carbon source and kan, velvets for replica plating minimal medium with glucose and kan with and without the amino acid, velvets for replica plating You may be able to think of many other phenotypes which could potentially be used in this experiment. Consult with your group and your TA before deciding on the phenotype you wish to obtain. Points to consider in designing your experiment. Will the mutation be lethal or be likely to show reduced growth? These possibilities may make it difficult or impossible to recover the desired mutant. How frequent is the mutant likely to be? If the desired mutant will only result from a mutation in a single gene, then you are OK if you can select for the mutant, but if you must screen for it, you will have to screen more than 10,000 mutants to be sure to find the one you want. Can the desired mutant phenotype result from a single mutation? For example, there are several pathways for methionine biosynthesis in A. tumefaciens and a mutation in one of them may not show a phenotype under most circumstances. You will need to spend some time on the biocyc web site http://biocyc.org/ looking up A. tumefaciens to be sure what you wish to try to do. Once you have decided on the mutation you wish to obtain, write up a protocol for the experiment and list all of the materials you will need. Show this protocol to the TA early in the afternoon. Consult with the TA as to which materials will be provided for you and which you will need to make. (For example, the materials required to purify the plasmid pRL27 will be provided for you, but plates containing various dyes will have to be made by you). Then make the media and solutions you will need. Pre-Laboratory Exercise Laboratory 8 part 2 1. Draw a map of the plasmid pRL27 indicating the antibiotic resistance genes and the ends of the transposon. 2. When you prepare plasmid DNA what happens to the chromosomal DNA during the preparation? 3. What happens to the RNA during the preparation? 4. What happens to proteins during the preparation? Laboratory 8 Part 2 Preparation of plasmid DNA You will be given fresh overnight cultures of E. coli containing the plasmid pRL27. The bacteria were grown in T broth with kanamycin. 1. Place 2 ml of each culture in a 2 ml Eppendorf tube. Label the tube. Pellet the cells in the centrifuge by spinning for 1 min. 2. Pour off the supernatant into disinfectant. Be sure to get rid of all of the supernatant. You can invert the tube and blot it on a Kinwipe to achieve this. Kill the bacteria on the Kimwipe with ethanol. Resuspend the bacterial pellet in 100 μl solution PI by vortexing very thoroughly. Solution PI. 3. 100 μg/ml RNase A 50 mM Tris-Cl pH 8.0 10 mM EDTA Add 100 μl solution P2. Mix by inverting (do not vortex). Incubate for 5 min. Solution P2. 0.2 M NaOH 1% SDS This solution must be made freshly shortly before use by mixing 800 μl water and 100 μl 2 M NaOH and 100 μl 10% SDS. Neither the NaOH or SDS solutions should be autoclaved although the water may be autoclaved to reduce nuclease activity. 4. Add 100 μl solution P3. Mix by inverting. Solution P3. 3 M K acetate adjusted to pH 5.0 with acetic acid 5. Centrifuge in Eppendorf, 10 min. 6. Transfer the aqueous phase to a new tube. Avoid the white precipitate which contains the chromosomal DNA (excluded from solution by high salt). 7. Add 0.7 volumes of isopropanol to precipitate the DNA. 8. Centrifuge in the Eppendorf centrifuge 10 min. 9. Remove the supernatant carefully using a micropipettor. Keep the pellet. Add 1 ml of 70% ethanol to wash the pellet. Mix by inverting. 10. Centrifuge in the Eppendorf centrifuge 10 min. 11. Remove the supernatant carefully using a micropipetter. Try to remove all of the ethanol using a blue and then a yellow tip. Remove as much ethanol as possible so that your pellet will dry in a reasonable time. 12. Give your tube labeled with your initials to your TA to be dried in a speed vac. 13. When the pellet has been dried, add 35 μl sterile water to the dried pellet. Mix stirring with the pipette tip. 14. Label the tube with your name and the name of the plasmid you prepared, use it for transposon mutagenesis and store the remainder in the refrigerator in case something goes wrong and you wish to repeat the mutagenesis. Introduction of the plasmid into Agrobacterium tumefaciens to obtain transposon mutants 1. You will be given a culture of A. tumefaciens C58 grown overnight in YEP and diluted and grown again in YEP until they reached mid-log phase. 2. Collect 2 ml of cells by centrifugation in the microfuge for 1 min. Pour off the supernatant. 3. Resuspend the cells in 0.1 ml of ice-cold sterile 20 mM CaCl2 and keep the cells on ice. 4. Add about 10 μl of plasmid DNA. 5. Freeze the cells in liquid nitrogen. 6. Thaw the cells by incubating them in a 37o water bath for 5 min. 7. Add 1 ml of YEP medium to the tube and incubate it overnight on a roller drum at room temperature. Save the transformed C58 in case you wish to make additional plates. Next day Plate 0.1 ml of your cells and 0.1 ml of a 1/10 dilution of your cells in 0.9% NaCl on medium containing kanamycin to obtain transposon mutants. The exact medium you use will depend on your experiment. Let the plated cells grow at room temperature for 3-7 days. Part 3 Characterize your mutant Following the protocol you wrote, characterize your mutants. Do they all seem to behave identically or are there various groups of mutants? Part 4 Finish characterizing your mutants and prepare stocks The day before the lab inoculate each of your cultures for overnight growth in liquid medium in a tube on the room temperature roller drum. 1. Finish the work contained in your protocol. 2. Make a stock of each of your mutants for use by either next year’s class or you in the next semester. To do this label a sterile Eppendorf clearly with the name of your mutant on both the top and side. Place 0.1 to0.2 ml of sterile glycerol in the tube. Add about 1 ml of your culture and invert the tube to mix the culture. Give the tube to your TA to be stored frozen at -80o. 3. Give your TA a detailed list of your mutants containing their names and properties to go with the stock cultures. Write a detailed report of your research in the format of a paper for The Journal of Bacteriology. Laboratory 8 Biology 422L Appendix 1: Notes on the preparation of plasmid DNA (Numbers refer to the numbers used in the protocol for the preparation of plasmid DNA above). 2. Solution P1 contains EDTA which will weaken the outer membrane and RNase which will digest the RNA released during cell lysis. 3. Solution P2 contains base to hydrolyze the cell walls and detergent to lyse the cell plasma membrane. 4. Solution P3 is very high in salt and buffer. It will nuetralize the base used to lyse the cells. The high salt will exclude the chromosomal DNA from solution. Appendix 2: Preparation of Competent E. coli Reference: D.A. Morrison (1979). In: Methods in Enzymology, Vol. 68; R. Wu, editor, page 326 (with minor adaptations). 1. Prepare a 5 ml ON in LB at appropriate temperature. 2. Inoculate 200 ml of LB + 10% glycerol with 2 ml ON culture and shake in a waterbath at appropriate temperature until Klett is about 65-75 (mid-log). 3. Immediately chill cells in a H2O-ice bath by swirling the flasks until cells are at 5o; transfer cells to sterile 250 ml ctg bottle. 4. Spin the culture at 6K for 10 min in large (GSA) rotor. 5. Gently resuspend cells with a cut-off pipet in 50 ml ice-cold 0.1 M MgCl2 and divide into 2 x 25 ml in 2 sterile, 30 ml, plastic ctg tubes. 6. Spin in an SS34 rotor at 6K for 10 min. 7. Resuspend the pellets in 25 ml ice-cold 0.1 M CaCl2 gently with a cut-off pipet. 8. Incubate cells on ice for 20 min. 9. Spin at 6K for 10 min. 10. Resuspend the pellets in a total of 8.6 ml of ice-cold 0.1 M CaCl2 gently and add 1.4 ml sterile 100% glycerol. Mix well with a cut-off pipet. 11. Transfer the 10 ml glycerinated, competent cells to small, sterile screw cap tubes (at f.e. 1 ml/tube) which are sitting in a H2O-ice bath. 12. Dry tubes and freeze immediately in a dry ice-ethanol bath for 5-10 min. 13. Transfer to the -80o freezer. Notes: All steps on ice from cell harvest on!! For use, thaw cells in a H2O-ice bath for 10 min - use as usual. Needed: 100% glycerol (sterile) LB 0.1 M MgCl2 (7.6) sterile 0.1 M CaCl2 (7.6) sterile Appendix 3: Gel Electrophoresis of Nucleic Acids Separation of DNA by gel electrophoresis depends on 1. The molecular weight of the DNA. Linear double-stranded DNA moves at a rate proportional to 1/log10 MW where MW = molecular weight. 2. The conformation of the DNA. The conditions under which the gel is run (buffer ionic strength, agarose concentration, ethidium bromide concentration and voltage) affect the relative mobilities of different forms. In general covalently closed circular DNA migrates the fastest with the rate of migration increasing with increasing supercoiling. Nicked circular DNA and linear DNA usually are relatively close in mobility with linear DNA running slightly faster than nicked circular DNA. As well as monomer plasmid molecules, you may observe dimers (supercoiled, nicked, and linear) in your DNA preparation. 3. The agarose concentration. The lower the agarose concentration the faster the DNA moves. 4. The applied current. As the current is increased the rate of migration of high molecular weight DNA increases faster than that of low molecular weight DNA. Best resolution of DNA is usually obtained at about 5 v/cm. The base composition of the DNA does not generally affect its mobility. RNA moves much faster than DNA in agarose gels.