Tutorial - iPlant Pods
advertisement

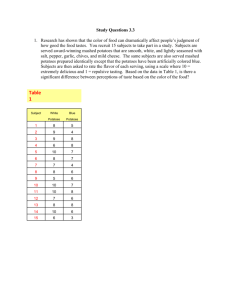
MAKER-P Genome Annotation using Atmosphere Rationale and background: MAKER-P is a flexible and scalable genome annotation pipeline that automates the many steps necessary for the detection of protein coding genes. MAKER-P identifies repeats, aligns ESTs and proteins to a genome, produces ab initio gene predictions, and automatically synthesizes these data into gene annotations having evidence-based quality indices. MAKER-P was developed by the Yandell Lab. Its predecessor, MAKER, is described in several publications (Cantarel et al. 2008; Holt & Yandell 2011). Additional background is available at the MAKER Tutorial at GMOD and is highly recommended reading. MAKER-P v2.28 is currently available as an Atmosphere image and is MPI-enabled for parallel processing. This tutorial will take users through steps of: 1. Launching the MAKER-P Atmosphere image 2. Setting up and running MAKER-P on an example small genome assembly 3. Copying output back to your iPlant Data Store using icommands 4. Visualizing MAKER-P output using the Integrated Genome Viewer (IGV) Note: Parts of this tutorial requires stand-alone VNC Viewer. Please download VNC Viewer (For windows, if you are unsure of which version to download, select the 32bit.exe file) Part 1: Connect to an instance of an Atmosphere Image (virtual machine) Step 1. Go to https://atmo.iplantcollaborative.org and log in with your iPlant credentials. Step 2. Click on the Launch New Instance button and search for MAKER-P_2.28 Step 3. Select the image MAKER-P_2.28 (emi-F13821D0) and click Launch Instance. It will take 10-15 minutes for the cloud instance to be launched. You will be notified by email when the image is ready. Note: Instances can be configured for different amounts of CPU, memory, and storage depending on user needs. This tutorial can be accomplished with the small instance size, m1.small (default). For real genome annotation larger instances will be required. Step 4. Launch the standalone VNC Viewer application on your desktop computer and enter your VM's IP address following by :1 Once you have connected, you will see your virtual Desktop, a Linux server running in the Atmosphere cloud computing platform. You will interact with it the same way as you would a physical computer. Part 2: Set up and run MAKER-P using the Terminal window Step 1. On the desktop click Terminal. A terminal window will open giving a command-line interface. Step 2. Get oriented. You will find staged example data in folder "MAKER-P_example_data" within the folder "Desktop". List its contents with the ls command: $ ls Desktop/MAKER-P_example_data/ README mRNA.fasta maker_exe.ctl example_output maker_bopts.ctl maker_opts.ctl msu-irgsp-proteins.fasta plant_repeats.fasta test_genome.fasta The fasta files include a scaled-down genome (test_genome.fasta) comprised of the first 100kb of each chromosome of rice. The remaining fasta files provide evidence for annotation: mRNA sequences from NCBI, publicly available annotated protein sequences of rice (MSU7.0 and IRGSP1.0), and a collection of plant repeats. The maker_*.ctl files are configuration files for MAKER which can be optionally used for this exercise or generated as described later. Executables for running MAKER-P are located in /opt/maker/bin and /opt/maker/exe: $ ls /opt/maker/bin cegma2zff fasta_merge chado2gff3 fasta_tool compare genemark_gtf2gff3 iprscan2gff3 iprscan_wrap maker maker2jbrowse maker2wap maker2zff maker_functional_gff maker_map_ids map2assembly map_gff_ids mpi_evaluator mpi_iprscan cufflinks2gff3 evaluator gff3_merge ipr_update_gff $ ls /opt/maker/exe/ RepeatMasker augustus blast maker2chado maker2eval_gtf exonerate maker_functional maker_functional_fasta mpich2-1.5 mpich2.tar.gz map_data_ids map_fasta_ids tophat2gff3 snap As the names suggest the /opt/maker/bin directory includes many useful auxiliary scripts. For example cufflinks2gff3 will convert output from an RNA-seq analysis into a GFF3 file that can be used for input as evidence for MAKER-P. Both Cufflinks and cufflinks2gff3 are available as tools the iPlant Discovery Environment (DE). Other auxiliary scripts now available in the DE include: tophat2gff3, maker2jbrowse, and maker2zff. RepeatMasker, Augustus, Blast, Exonerate, and SNAP are programs that MAKER-P uses in its pipeline. We recommend the MAKER Tutorial at GMOD for additional reading about these. Step 3. Set up your environment. Note: this step is not essential for the purpose of this tutorial. Skipping this step you will need to specify the complete path when running the maker command (/opt/maker/bin/maker). $ export PATH=/opt/maker/bin/:$PATH $ export ZOE=/opt/maker/exe/snap $ export AUGUSTUS_CONFIG_PATH=/opt/maker/exe/augustus/config Step 4. Set up a MAKER-P run. Create a working directory called "maker_run" using the mkdir command and use cd to move into that directory: $ mkdir maker_run $ cd maker_run Step 5. Create another directory called "test_data" and copy the staged fasta data into test_data using cp. Verify using the ls command $ cp ~/Desktop/MAKER-P_example_data/*.fasta test_data/. $ ls test_data $ ls test_data/ mRNA.fasta msu-irgsp-proteins.fasta plant_repeats.fasta test_genome.fasta Step 6. Run the maker command with the --help flag to get a usage statement and list of options: $ maker --help MAKER version 2.28 Usage: maker [options] <maker_opts> <maker_bopts> <maker_exe> Description: MAKER is a program that produces gene annotations in GFF3 format using evidence such as EST alignments and protein homology. MAKER can be used to produce gene annotations for new genomes as well as update annotations from existing genome databases. The three input arguments are control files that specify how MAKER should behave. All options for MAKER should be set in the control files, but a few can also be set on the command line. Command line options provide a convenient machanism to override commonly altered control file values. MAKER will automatically search for the control files in the current working directory if they are not specified on the command line. Input files listed in the control options files must be in fasta format unless otherwise specified. Please see MAKER documentation to learn more about control file configuration. MAKER will automatically try and locate the user control files in the current working directory if these arguments are not supplied when initializing MAKER. It is important to note that MAKER does not try and recalculated data that it has already calculated. For example, if you run an analysis twice on the same dataset you will notice that MAKER does not rerun any of the BLAST analyses, but instead uses the blast analyses stored from the previous run. To force MAKER to rerun all analyses, use the -f flag. MAKER also supports parallelization via MPI on computer clusters. Just launch MAKER via mpiexec (i.e. mpiexec -n 40 maker). MPI support must be configured during the MAKER installation process for this to work though Options: -genome|g <file> -RM_off|R -datastore/ nodatastore -old_struct -base <string> -tries|t <integer> -cpus|c <integer> -force|f -again|a -quiet|q -qq -dsindex Overrides the genome file path in the control files Turns all repeat masking options off. Forcably turn on/off MAKER's two deep directory structure for output. Always on by default. Use the old directory styles (MAKER 2.26 and lower) Set the base name MAKER uses to save output files. MAKER uses the input genome file name by default. Run contigs up to the specified number of tries. Tells how many cpus to use for BLAST analysis. Note: this is for BLAST and not for MPI! Forces MAKER to delete old files before running again. This will require all blast analyses to be rerun. recaculate all annotations and output files even if no settings have changed. Does not delete old analyses. Regular quiet. Only a handlful of status messages. Even more quit. There are no status messages. Quickly generate datastore index file. Note that this -nolock -TMP -CTL -OPTS -BOPTS -EXE -MWAS <option> will not check if run settings have changed on contigs Turn off file locks. May be usful on some file systems, but can cause race conditions if running in parallel. Specify temporary directory to use. Generate empty control files in the current directory. Generates just the maker_opts.ctl file. Generates just the maker_bopts.ctl file. Generates just the maker_exe.ctl file. Easy way to control mwas_server for web-based GUI options: STOP START RESTART -version Prints the MAKER version. -help|? Prints this usage statement. Step 7. Create control files that tell MAKER-P what to do. Three files are required: • maker_opts.ctl - gives location of input files (genome and evidence) and sets options that affect MAKER-P behavior • maker_exe.ctl - gives path information for the underlying executables. • maker_bopt.ctl - sets parameters for filtering BLAST and Exonerate alignment results To create these files run the maker command with the -CTL flag. Verify with ls: $ maker -CTL $ ls maker_bopts.ctl maker_exe.ctl maker_opts.ctl test_data The maker_exe.ctl is automatically generated with the correct paths to executables and does not need to be modified. The maker_bopt.ctl is automatically generated with reasonable default parameters and also does not need to be modified unless you want to experiment with optimization of these parameters. The automatically generated maker_opts.ctl file does need to be modified in order to specify the genome file and evidence files to be used as input. Several text editors are available, including emacs, nano and gedit. If you have not tried any of these gedit probably is easiest to learn as it has a familiar graphical user interface. It can be started by typing gedit & on the command line or by selecting it from the menu on the VNC desktop (Applications=>Accessories=>Text Editor). Note: If pressed for time a pre-edited version of the maker_opts.ctl file is staged in ~/Desktop/MAKER-P_example_data/. Delete the current file and copy the staged version here. Then skip to Step 8. $ rm maker_opts.ctl $ cp ~/Desktop/MAKER-P_example_data/maker_opts.ctl . Here are the sections of the maker_opts.ctl file you need to edit. Add path information to files as shown. Do not allow any spaces after the equal sign or anywhere else. You can specify a complete path or relative path as shown here. In general you can specify multiple files by separating with a comma without spaces. This section pertains to specifying the genome assembly to be annotated and setting organism type: #-----Genome (these are always required) genome=./test_data/test_genome.fasta #genome sequence (fasta file or fasta embeded in GFF3 file) organism_type=eukaryotic #eukaryotic or prokaryotic. Default is eukaryotic The following section pertains to EST and other mRNA expression evidence. Here we are only using same species data, but one could specify data from a related species using the altest parameter. With RNA-seq data aligned to your genome by Cufflinks or Tophat one could use maker auxiliary scripts (cufflinks2gff3 and tophat2gff3) to generate GFF3 files and specify these using the est_gff parameter: #-----EST Evidence (for best results provide a file for at least one) est=./test_data/mRNA.fasta #set of ESTs or assembled mRNA-seq in fasta format altest= #EST/cDNA sequence file in fasta format from an alternate organism est_gff= #aligned ESTs or mRNA-seq from an external GFF3 file altest_gff= #aligned ESTs from a closly relate species in GFF3 format The following section pertains to protein sequence evidence. Here we are using previously annotated protein sequences. Another option would be to use SwissProt or other database: #-----Protein Homology Evidence (for best results provide a file for at least one) protein=./test_data/msu-irgsp-proteins.fasta #protein sequence file in fasta format (i.e. from mutiple oransi protein_gff= #aligned protein homology evidence from an external GFF3 file This next section pertains to repeat identification: #-----Repeat Masking (leave values blank to skip repeat masking) model_org= #select a model organism for RepBase masking in RepeatMasker rmlib=./test_data/plant_repeats.fasta #provide an organism specific repeat library in fasta format for RepeatM repeat_protein= #provide a fasta file of transposable element proteins for RepeatRunner rm_gff= #pre-identified repeat elements from an external GFF3 file prok_rm=0 #forces MAKER to repeatmask prokaryotes (no reason to change this), 1 = yes, 0 = no softmask=1 #use soft-masking rather than hard-masking in BLAST (i.e. seg and dust filtering) Various programs for ab initio gene prediction can be specified in the next section. Here we are using SNAP set to use an HMM trained on rice. Specifying the entire path to the hmm file would not be necessary if the ZOE environment variable is set as described in step 3 above. #-----Gene Prediction snaphmm=/opt/maker/exe/snap/HMM/O.sativa.hmm #SNAP HMM file gmhmm= #GeneMark HMM file augustus_species= #Augustus gene prediction species model fgenesh_par_file= #FGENESH parameter file pred_gff= #ab-initio predictions from an external GFF3 file model_gff= #annotated gene models from an external GFF3 file est2genome=0 #infer gene predictions directly from ESTs, 1 = protein2genome=0 #infer predictions from protein homology, 1 unmask=0 #also run ab-initio prediction programs on unmasked (annotation pass-through) yes, 0 = no = yes, 0 = no sequence, 1 = yes, 0 = no These lines control other behaviors of MAKER-P and do not need to be changed for this demonstration: #-----Other Annotation Feature Types (features MAKER doesn't recognize) other_gff= #extra features to pass-through to final MAKER generated GFF3 file #-----External Application Behavior Options alt_peptide=C #amino acid used to replace non-standard amino acids in BLAST databases cpus=1 #max number of cpus to use in BLAST and RepeatMasker (not for MPI, leave 1 when using MPI) #-----MAKER Behavior Options max_dna_len=100000 #length for dividing up contigs into chunks (increases/decreases memory usage) min_contig=1 #skip genome contigs below this length (under 10kb are often useless) pred_flank=200 #flank for extending evidence clusters sent to gene predictors pred_stats=1 #report AED and QI statistics for all predictions as well as models AED_threshold=1 #Maximum Annotation Edit Distance allowed (bound by 0 and 1) min_protein=0 #require at least this many amino acids in predicted proteins alt_splice=0 #Take extra steps to try and find alternative splicing, 1 = yes, 0 = no always_complete=1 #extra steps to force start and stop codons, 1 = yes, 0 = no map_forward=0 #map names and attributes forward from old GFF3 genes, 1 = yes, 0 = no keep_preds=0 #Concordance threshold to add unsupported gene prediction (bound by 0 and 1) split_hit=10000 #length for the splitting of hits (expected max intron size for evidence alignments) single_exon=0 #consider single exon EST evidence when generating annotations, 1 = yes, 0 = no single_length=250 #min length required for single exon ESTs if 'single_exon is enabled' correct_est_fusion=0 #limits use of ESTs in annotation to avoid fusion genes tries=3 #number of times to try a contig if there is a failure for some reason clean_try=0 #remove all data from previous run before retrying, 1 = yes, 0 = no clean_up=0 #removes theVoid directory with individual analysis files, 1 = yes, 0 = no TMP=/tmp #specify a directory other than the system default temporary directory for temporary files Step 8. Run MAKER-P Make sure you are in the maker_run directory and all of your files are in place. Perform these steps to check: $ pwd /home/steinj/maker_run $ ls maker_bopts.ctl maker_exe.ctl maker_opts.ctl stderr test_data $ ls test_data/ mRNA.fasta msu-irgsp-proteins.fasta plant_repeats.fasta test_genome.fasta Starting the MAKER-P run is as simple as entering the command maker. If you did not set the PATH environmental variable in Step 3 then you will need to type the entire path: /opt/maker/bin/maker. Because your maker control files are in the present directory you do not have to explicitly specify these in the command; they will be found automatically. MAKER-P automatically outputs thousands of lines of STDERR, reporting on its progress and producing warnings and errors if they arise. It is a good practice to redirect this output to a log file so you have a record of it, especially if something goes wrong. Since we are in the bash shell you can redirect the output by typing the command as follows: maker 2> log_file &. Another useful practice is to employ the unix time command to report statistics on how long the run took to complete. The output of time will appear at the end of the captured standard error file. Putting all of this together we type the following command to start MAKER-P: (time /opt/maker/bin/maker) 2> log_file & This part of the tutorial usually takes about 30 minutes to complete. You will know MAKER-P is finished when the log_file announces "Maker is now finished!!!". Use the tail command to look at the last 10 lines of the log_file: $ tail log_file processing chunk output processing contig output Maker is now finished!!! real user sys 31m11.250s 36m52.210s 1m34.106s Step 9. Examining MAKER-P output Output data appears in a new directory called test_genome.maker.output. Move to that directory and examine its contents. Note if your run is not yet complete move instead to the staged example output (cd ~/Desktop/MAKERP_example_data/example_output/test_genome.maker.output/): $ cd test_genome.maker.output/ $ ls maker_bopts.log maker_exe.log maker_opts.log mpi_blastdb seen.dbm test_genome.all.gff test_genome_datastore test_genome.db test_genome_master_datastore_index.log • The maker_opts.log, maker_exe.log, and maker_bopts.log files are logs of the control files used for this run of MAKER. • The mpi_blastdb directory contains FASTA indexes and BLAST database files created from the input EST, protein, and repeat databases. • test_genome_master_datastore_index.log contains information on both the run status of individual contigs and information on where individual contig data is stored. • The test_genome_datastore directory contains a set of subfolders, each containing the final MAKER output for individual contigs from the genomic fasta file. Check the test_genome_master_datastore_index.log to see if there were any failures: $ less test_genome_master_datastore_index.log Chr1 Chr1 Chr10 Chr10 Chr11 Chr11 Chr12 Chr12 Chr2 Chr2 Chr3 Chr3 Chr4 Chr4 Chr5 Chr5 Chr6 Chr6 Chr7 Chr7 Chr8 test_genome_datastore/41/30/Chr1/ test_genome_datastore/41/30/Chr1/ test_genome_datastore/7C/72/Chr10/ test_genome_datastore/7C/72/Chr10/ test_genome_datastore/1E/AA/Chr11/ test_genome_datastore/1E/AA/Chr11/ test_genome_datastore/1B/FA/Chr12/ test_genome_datastore/1B/FA/Chr12/ test_genome_datastore/E9/36/Chr2/ test_genome_datastore/E9/36/Chr2/ test_genome_datastore/CC/EF/Chr3/ test_genome_datastore/CC/EF/Chr3/ test_genome_datastore/A3/11/Chr4/ test_genome_datastore/A3/11/Chr4/ test_genome_datastore/8A/9B/Chr5/ test_genome_datastore/8A/9B/Chr5/ test_genome_datastore/13/44/Chr6/ test_genome_datastore/13/44/Chr6/ test_genome_datastore/91/B7/Chr7/ test_genome_datastore/91/B7/Chr7/ test_genome_datastore/9A/9E/Chr8/ STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED FINISHED STARTED Chr8 Chr9 Chr9 test_genome_datastore/9A/9E/Chr8/ test_genome_datastore/87/90/Chr9/ test_genome_datastore/87/90/Chr9/ FINISHED STARTED FINISHED All completed. Other possible status entries include: • FAILED - indicates a failed run on this contig, MAKER will retry these • RETRY - indicates that MAKER is retrying a contig that failed • SKIPPED_SMALL - indicates the contig was too short to annotate (minimum contig length is specified in maker_opt.ctl) • DIED_SKIPPED_PERMANENT - indicates a failed contig that MAKER will not attempt to retry (number of times to retry a contig is specified in maker_opt.ctl) The actual output data is stored in in nested set of directories under* test_genome_datastore* in a nested directory structure. A typical set of outputs for a contig looks like this: $ ls test_genome_datastore/41/30/Chr1/ Chr1.gff Chr1.maker.non_overlapping_ab_initio.proteins.fasta Chr1.maker.non_overlapping_ab_initio.transcripts.fasta Chr1.maker.proteins.fasta Chr1.maker.snap_masked.proteins.fasta Chr1.maker.snap_masked.transcripts.fasta Chr1.maker.transcripts.fasta run.log theVoid.Chr1 • The Chr1.gff file is in GFF3 format and contains the maker gene models and underlying evidence such as repeat regions, alignment data, and ab initio gene predictions, as well as fasta sequence. Having all of these data in one file is important to enable visualization of the called gene models and underlying evidence, especially using tools like Apollo which enable manual editing and curation of gene models. • The fasta files Chr1.maker.proteins.fasta and Chr1.maker.transcripts.fasta contain the protein and transcript sequences for the final MAKER-P gene calls. • The Chr1.maker.non_overlapping_ab_initio.proteins.fasta and Chr1.maker.non_overlapping_ab_initio.transcripts.fasta files are models that don't overlap MAKER-P genes that were rejected for lack of support. • The Chr1.maker.snap_masked.proteins.fasta and Chr1.maker.snap_masked.transcript.fasta are the initial SNAP predicted models not further processed by MAKER-P The output directory theVoid.Chr1 contains raw output data from all of the pipeline steps. One useful file found here is the repeat-masked version of the contig, query.masked.fasta. Step 10. Consolidate output using utility scripts fasta_merge and gff3_merge. Before running make sure you are in the same directory with the test_genome_master_datastore_index.log file. $ /opt/maker/bin/gff3_merge -d test_genome_master_datastore_index.log $ /opt/maker/bin/fasta_merge -d test_genome_master_datastore_index.log This results in the following new files. Note that the fasta files were merged in a fashion that maintains categories. test_genome.all.gff test_genome.all.maker.non_overlapping_ab_initio.proteins.fasta test_genome.all.maker.non_overlapping_ab_initio.transcripts.fasta test_genome.all.maker.proteins.fasta test_genome.all.maker.snap_masked.proteins.fasta test_genome.all.maker.snap_masked.transcripts.fasta test_genome.all.maker.transcripts.fasta Step 11. Copy data back to your iPlant Data Store. We will use icommands for this. First you will need to initialize icommands using the iinit command. Provide the information shown in red: $ iinit One or more fields in your iRODS environment file (.irodsEnv) are missing; please enter them. Enter the host name (DNS) of the server to connect to:data.iplantcollaborative.org Enter the port number:1247 Enter your irods user name:<your user name> Enter your irods zone:iplant Those values will be added to your environment file (for use by other i-commands) if the login succeeds. Enter your current iRODS password:<your iplant password> Typing ils will show the files and directories in your Data Store—give it a try. Create a folder for your data called “maker_atmosphere_tutorial_output” and move to that folder as follows: $ imkdir maker_atmosphere_tutorial_output $ icd maker_atmosphere_tutorial_output $ ils /iplant/home/steinj/maker_atmosphere_tutorial_output: Copy files to your Data Store using iput. For example: $ iput test_genome.all.gff $ iput test_genome.all.maker.proteins.fasta $ iput test_genome.all.maker.transcripts.fasta $ ils /iplant/home/steinj/maker_atmosphere_tutorial_output: test_genome.all.gff test_genome.all.maker.proteins.fasta test_genome.all.maker.transcripts.fasta For more on icommands… See https://pods.iplantcollaborative.org/wiki/display/start/Using+icommands Part 3: Visualize annotation data using the Integrated Genome Viewer (IGV): The MAKER-P Atmosphere image is loaded with several software packages for viewing genomics data. On the VNC Desktop open the folder NGS_Viewers to access and select IGV-2.0.7 The IGV will likely open with a pre-loaded genome, such as human. To load your genome go under the file menu and select Import Genome … Next use the dialog box to enter the genome assembly fasta “test_genome.fasta” and gene file “test_genome.all.gff”. Once loaded you can select a chromosome using the drop-down menus. By default the gene track is collapsed. To expand, right click on the track name and select expanded. Because the GFF3 files contain all of the evidence features in addition to genes so to will your track show data not limited to genes. This could be changed by parsing the GFF3 file into individual files containing individual features, e.g. repeat data and maker gene features.