emi412373-sup-0002-si
advertisement

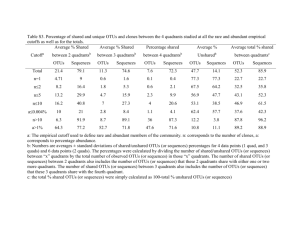
Supporting information Study Sites & Sample Collection Estuarine samples were taken from the Sydney harbour estuary (Port Jackson), which connects the inland Paramatta River with the Tasman Sea. The system receives diverse water inputs from inland tributaries, terrestrial run-off and storm water and contains strong salinity and nutrient gradients (Supplementary Table S1. Samples were collected along a transect spanning sites at the Parramatta river (33.8°S, 151.0 °W), Sydney Heads (33.8°S 151.3°W), and Middle Harbour (33.7°S, 151.2° W) which incorporated salinities ranging from 12.28 ppt to 33.3 ppt (Supplementary Table S1). Coastal and open ocean samples were collected during an oceanographic voyage aboard the RV Southern Surveyor in October 2010 (Supplementary Figure S2). Coastal samples covered an area of the Tasman sea between 28°S-33°S within the continental shelf, and included samples collected from three river plume regions including the Richmond, Clarence and Macleay river. Open ocean samples were collected across a region spanning 29°S -32°S and incorporated several oceanographic features including the East Australia Current and localized cold core eddies. At all locations, 2 L water samples were collected from surface waters. For the coastal and open water samples, physicochemical properties of the water column were measured with a Seabird SBE911 CTD and water samples were collected using 10 L Niskin bottles. Estuarine samples were collected in acid washed and triple rinsed poly propylene bottles. Estuarine physicochemical properties were measured with a multiparameter water quality CTD (YSITM). Samples for estuarine nutrient determination were filtered at the time of collection (0.45μm) analysed using flow injection analysis (LaChatTM). Between collection and analysis nutrient samples were kept frozen (-30°C) for not more than 3 months. Samples for collection of microbial DNA were filtered onto 0.2µm polycarbonate filters (Millipore) and immediately frozen. Total DNA was subsequently extracted using bead beating and chemical lysis (Powerwater DNA isolation kit, MoBio, USA) with an additional heating step at 70°C (10 minutes) to maximize fungal lysis. ITS Sequencing The fungal ITS region was amplified from community DNA using the ITS1F primer (3’CTTGGTCATTTAGAGGAAGTAA-5’) (Gardes and Bruns, 1993) and ITS4 primer (3’TCCTCCGCTTATTGATATGC-5’) using the following reaction conditions: a single-step 30 cycle PCR using HotStarTaq Plus Master Mix Kit (Qiagen) comprising 94 °C initial denaturation (3 min), 28 cycles of 94 °C (30 sec) 53 °C (40 sec) and 72 °C (1 min), with a final elongation step at 72 °C (5 min). The resultant amplicons were purified using Agencourt Ampure beads (Agencourt Bioscience Corporation, USA) and sequenced using Roche 454 FLX Titanium (Molecular Research LP, Texas) instruments and reagents following the manufacturers guidelines (Dowd et al., 2008). Bioinformatics Samples were de-multiplexed and processed using the QIIME package (Caporaso et al., 2010). After sequencing, barcodes and primers were removed from the sequences, sequences shorter than 200bp were removed, sequences with ambiguous base calls were removed, and sequences with homopolymer runs exceeding 6 bp were removed. A 200bp cut-off was chosen as this is a sequence quality related metric incorporated in the de-multiplexing and quality control step of the QIIME pipeline (http://qiime.org/scripts/split_libraries.html). Despite the potential that read-trimming may introduce a taxonomic bias for those organisms with very short ITS regions, several other studies have used cut-offs of <200bp (e.g. De Beek et al., 2014; Bokulich and Mills, 2013) or < 300bp (Amend et al., 2010) with no apparent bias. Using a 200bp cut-off should have little taxonomic bias as ITS regions amplified with the ITS1/ITS4 primer set span a region that ranges in size between 400bp and 800bp (Bellermain et al., 2010). Additionally, despite the heterogeneity in length, the average size of the full ITS region is 600bp across all fungal divisions (ODe Beek et al., 2014). Operational taxonomic units (OTUs) were defined as sequences sharing 97% similarity (Buée et al., 2009) using UCLUST (Edgar, 2010). Additionally, fungal ITS1 regions were extracted from the full length amplicons using ITSx (Bengtsson‐ Palme, et al 2013) and analysed as above to account for the potential influence of length heterogeneity. This analysis yielded fewer O.T.Us (7956 O.T.Us) compared to full length amplicons (19260 O.T.Us) however beta-diversity patterns were maintained in this dataset (Supporting Information Figure S4) indicating the ITS region used does not greatly influence the partitioning of habitats in this dataset. Overall, 97.2% of the sequences analysed contained fungal ITS1 regions identified using fungi specific Hidden Markov Models (Bengtsson‐ Palme, et al 2013) indicating that the diversity we profiled was indeed fungal. Samples were rarefied to 1082 sequences to ensure even sampling depth. Representative sequences of each OU were compared against the UNITE database (http://unite.ut.ee/index.php) (Kõljalg et al., 2005) using BLAST (E<105 ) (Altschul et al., 1991) to assign taxonomy. Statistical analysis using multivariate and network analysis OTU abundance data was square root transformed and Bray-Curtis similarity between profiles was ordinated using multidimensional scaling (MDS), while Analysis of Similarities (ANOSIM) was applied to determine if differences in factors (habitats) were significantly different from a randomly permutated distribution. Distance based multivariate analysis for linear models using Primer 6 (version 6.1.11, Primer-E, Clarke and Gorley, 2006), which describes the relationship between multivariate analysis and environmental variables based on a resemblance matrix using permutations was employed to link biotic patterns to environmental variables. The environmental matrix (Euclidean distance) was generated using available metadata (supporting table S1) with the exception of latitude and longitude and those variables for which insufficient data was collected across the whole dataset (TDN,TDP,TN,TP,POP,DON,DOP,POC) and excluding those samples for which insufficient environmental data was collected (7 samples). Values which were below the detection limit were treated as zeroes. The OTU network was generated in QIIME using the make_otu_network.py python script (Caporaso et al., 2010, http://qiime.org/scripts/make_otu_network.html) and visualized using Cytoscape (version 3.0.1, National resource for network biology, Shannon et al., 2003). Nodes in the bi-partite network represented both samples and individual OTUs. Edges connect OTUs to the samples in which they occur with the samples clustering based on the degree to which they share OTUs. Edge weights were determined using the abundance of individual OTUs in each sample. A stochastic spring-embedded layout was utilized in Cytoscape (“Edge-Weighted Spring Embedded”) in which nodes act like physical objects that repel each other, and edges act a springs with a spring constant and a resting length. Nodes are organized in a way that minimizes forces in the network bringing together samples that share the most OTUs, scaled by abundance. Supplementary references Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nuc Acid Res 25: 3389-3402 Amend, A. S., Seifert, K. A., & Bruns, T. D. (2010). Quantifying microbial communities with 454 pyrosequencing: does read abundance count?.Molecular Ecology, 19(24), 5555-5565. Bellemain, E., Carlsen, T., Brochmann, C., Coissac, E., Taberlet, P., & Kauserud, H. (2010). ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. Bmc Microbiology, 10(1), 189. Bengtsson‐ Palme, Johan, et al. (2013) mproved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data." Methods in Ecology and Evolution 4: 914-919. Bokulich, N. A., & Mills, D. A. (2013). Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Applied and environmental microbiology, 79(8), 2519-2526. Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S, Martin F (2009). 454pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184: 449-456 De Beeck, M. O., Lievens, B., Busschaert, P., Declerck, S., Vangronsveld, J., & Colpaert, J. V. (2014). Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies, PLoS One, e97629 Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. (2008). Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8: 43 Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010a). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336. Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460-2461. Clarke KR, Gorley RN (2006). PRIMER v6: User Manual/Tutorial. PRIMER-E, Plymouth. Gardes M, Bruns, TD (1993). ITS primers with enhanced specificity for basidiomycetes application to the identification of mycorrhizae and rusts. Mol Ecol 2: 113-118. Kõljalg U, Larsson KH, Abarenkov K, Milsson RH, Alexander IJ, Eberhardt U, Erland S, Hølland F, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vrålstad T, Ursing BM (2005). UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166: 1063-1068 Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowshi B, Ideker T (2003). Cytoscape: A software environment for integrated models of biomedical interaction networks. Gen Res 13: 2498-2504. Supporting information Figure S1. Distance based linear model showing correlation of environmental variables to 454-sequencing data defined at 97% sequence identity (Estuary , Coastal and Ocean ) Supporting information Figure S2. Map of sampling sites from Sydney Harbour Estuary (Panel A) and Coastal and Ocean sites (Panel B, CH=coast-harbour (range), CR=coast-river (Yellow), C=coast (Green), STS=South Tasman Sea (maroon), TS=Tasman Sea (pink), EAC=East Australian current (Brown), CCE=Cold core eddy (Blue)) Supporting information figure S3. Analysis using extracted ITS1 regions. A) Multidimensional scaling of relative OTU abundance (Estuary , Coastal and Ocean ),and (B) Network analysis of OTU partitioning between samples. Sample clustering represents the degree to which OTUs are shared between samples, with samples grouping closer together if they share more OTUs. Black nodes represent OTUs, coloured nodes represent sample type (red = estuary; green = coastal; blue = open ocean) and edges (lines) connect OTUs to samples in which they occur (coloured by the habitat of the connected sample). Edge weights represent the number of sequences from each OTU that occurred in each sample. ITS1 OTUs were clustered at 97% identity using UCLUST (Edgar, 2010). Networks were generated using QIIME (Caporaso et al 2010) and visualized using Cytoscape (Shannon et al, 2003 ). All samples were rarefied to 1045 sequences per sample to equalize sampling depth. Supplementary Fig. S1 A B Supplementary Fig. S2 Supplementary Fig. S3