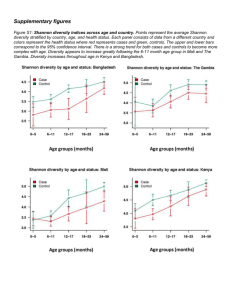
emi412282-sup-0003-si
advertisement

SI text 1: Automated Intergenic Spacer Analysis (ARISA) and sequence processing workflows. Sequencing was performed at Research and Testing Laboratories (Lubbock, Texas, USA). For the bacteria the V4-V6 hypervariable region of the 16S gene was sequenced on the Roche 454 FLX+ system (Roche, Basel, Switzerland); for the eukaryotes the V4 hypervariable region of the 18S rRNA gene was sequenced on the Illumina MiSeq platform. Raw sequences were denoised (only 454 sequencing data) and quality trimmed in mothur v.1.33.3 (Schloss et al. 2009) and submitted to SILVAngs (Quast et al. 2013) for taxonomic classification. Clustering of OTUs was performed at 97% sequence identity and taxonomic classification at 93% sequence identity on genus level. Sequence counts were adjusted to the minimum number of sequences per sample (454 sequencing: 5249, Illumina sequencing: 19474). Single OTU occurrences and sequence contaminations from animals and E. acroides were removed prior to the analysis of the eukaryotic dataset (Logares et al. 2014). Sample C1 was dominated by OTUs assigned to an annelid worm. After the removal of contaminating sequences, OTU richness in sample C1 was much lower than in the other samples presumably due to the compositional character of DNA samples, i.e. because of the presence of a highly dominant OTU, DNA belonging to rare organisms was less likely to be sequenced than in samples with a more even distribution. The larger number of eukaryotic OTUs in comparison to the bacterial dataset was due to the increased sequencing effort with the Illumina technology. After rarefying the eukaryotic sequences to the minimum of the bacterial sequence counts and accounting for the large proportion of rare bacterial OTUs, i.e. comparing the number of eukaryotic OTUs to the Chao1 estimated bacterial OTU richness, there were approximately twice as many bacterial as eukaryotic OTUs. The eukaryotic community data was further converted to presence/absence to account for multicellularity and variations in rRNA gene copy number per genome (Prokopowich et al. 2003; Logares et al. 2014). SI references: Logares, R. et al., 2014. Patterns of rare and abundant marine microbial eukaryotes. Current biology : CB, 24(8), pp.813–821. Prokopowich, C.D., Gregory, T.R. & Crease, T.J., 2003. The correlation between rDNA copy number and genome size in eukaryotes. Genome, 46, pp.48–50. Quast, C. et al., 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research, 41(Database issue), pp.D590–6. Schloss, P.D. et al., 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology, 75(23), pp.7537–41.