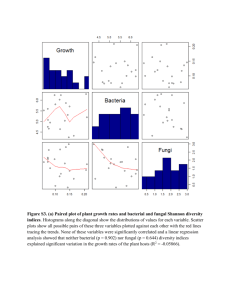
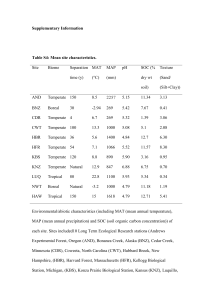
supplementary methods
advertisement

SUPPLEMENTARY METHODS Multiplex 454 pyrosequencing data processing Multiplex tag pyrosequencing was performed by the Research and Testing Laboratory (Lubbock, Texas, USA) using the 454 FLX titanium platform (Roche, Branford, CT, USA). Bacterial and fungal assemblages were taxonomically characterised by sequencing of the bacterial 16S rRNA genes amplified using primers 28F and 519R (Quere et al. 2005), and the fungal ITS region using primers ITS1 (Gardes and Bruns 1993) and ITS4R (White et al. 1990) respectively. Sequence data were analysed using the MOTHUR pipeline (Schloss et al. 2009). Raw data were provided as standard flowgram format files, and were extracted and error-checked via the Pyronoise algorithm (Quince et al. 2011). Sequence data were further quality screened by removing short reads (<150 bp), long homopolymers (>8 repeats) and truncation of 16S reads (>450 bp). The sequence data was checked for chimeras using UCHIME algorithm (Edgar et al. 2011), and then were preclustered at 1% to account for 454’s titanium instrument error rate. Datasets were initially subsampled to equal depth; however, after determining no significant impact between sub-sampled and non-subsampled datasets, the dataset was left intact. Bacterial sequences were aligned to the curated SILVA secondary structure alignment (Pruesse et al. 2007), and then clustered into operational taxonomic units (OTUs) based on 96% sequence similarity (Kim et al. 2011). The taxonomic position of identified bacterial OTUs was assigned using the Greengenes database (2013 May version) ((McDonald et al. 2012, Werner et al. 2012)) that was trimmed to the same region as the amplicons (V1-V3). An OTU abundance-by sample matrix was generated from the bacterial dataset, and then the singletons were removed from sample matrix. For fungal amplicons, after the ITS1 region was extracted from the amplicon using software package developed by Nilsson (in press), USEARCH software package (Edgar 2010) was used to cluster fungal ITS1 sequences at 97% sequence similarity to loosely define OTUs. The representative sequence were picked by MATTF v6 (Katoh and Toh 2008), and they were then compared against UNITE fungal ITS database (Koljalg et al. 2005) by BLASTn (Altschul et al. 1990), and the top 15 matches were retrieved. To correct the clustering error, OTUs that shared over 50% identical top matches were grouped together manually. The resulting OTU abundance matrix was generated from the fungal community data with QIIME package (Caporaso et al. 2010). Reference Caporaso JG et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335-336 Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460-2461 Edgar RC (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194-2200 Altschul SF et al (1990) Basic local alignment search tool. J. Mol. Biol. 215(3):403-410 Gardes M and Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes‐ application to the identification of mycorrhizae and rusts. Microb Ecol 2(2):113-118 Koljalg U et al (2005) UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166:1063-1068 Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286-298 McDonald D et al (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610-618 Nilsson RH et al (in press) An ITS1/ITS2 extractor for the fungal ITS region. Fungal Ecology Pruesse E et al (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35:7188-7196 Kim M et al (2011) Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J Microbiol Methods 84:81-87 Quince C et al (2011) Removing noise from pyrosequenced amplicons." BMC Bioinform 12(1): 38 Quere CL et al (2005) Ecosystem dynamics based on plankton functional types for global ocean biogeochemistry models. Glob Chang Biol 11(11):2016-2040 Schloss PD et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537-7541. doi:10.1128/aem.01541-09 Werner JJ et al (2012) Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6:94-103 White TJ et al (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications 18:315-322