nph12758-sup-0001-Supportinginformation
advertisement

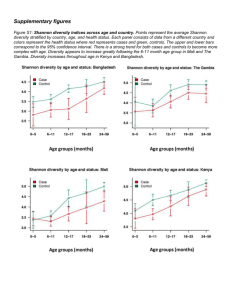
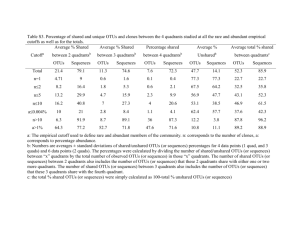
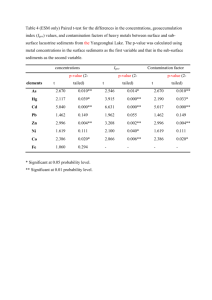
Supporting Information Methods S1, Tables S1–S3 and Figs S1–S5 Methods S1 MCA OTU delineation Quality-filtered sequence data was pre-clustered at 97% identity using CD-HIT. Then, a multiple sequence alignment of cluster centroid sequences was performed using the program MAFFT, version 6.925 (Katoh & Standley, 2013), with the FFT-NS-2 strategy assuming multiple conserved regions and long gaps. This setting works with large data sets. The aligned data set was then split into subsets based on branching patterns and reproducibility of the clusters for more detailed analyses. One or two groups often dominate, and these dominant sequence types were analyzed separately. The new datasets were aligned again with MAFFT, and each of the data sets was clustered using a neighbor net analysis (Huson et al., 2011). Neighbor net is an algorithm for constructing phylogenetic networks and is based on the neighbor joining method. Similar to neighbor joining, the method uses a distance matrix as input and agglomerating clustering. The neighbor net algorithm can make overlapping clusters, which do not form a hierarchy. The result is presented as a phylogenetic network referred to as a split network. Monophyletic clades (OTUs) were identified in these split networks as terminal clades without reticulations (see example of a reticulation in Fig. S1b). From these terminal clades representative sequences were selected to construct a neighbor net or a phylogenetic tree to illustrate the distance between the OTUs. OTUs represented by only one (Site and Regional) or fewer than five (Local) sequence(s) in the data set were removed prior to analysis. 97% OTU delineation using QIIME All sequence analysis was done using QIIME 1.6.0 (Caporaso et al., 2010a; QIIME script available upon request) using the same sequences as in the MCA clustering. OTUs were picked de novo using 97% similarity (90-99% similarity was used for the supervised learning approach), using the UCLUST algorithm (QIIME script: pick_otus.py) according to Edgar (2010). Cluster centroids for OTUs were chosen as the OTU representative sequences. Abundance matrices of OTUs across sample locations (OTU tables) were constructed, and OTUs represented by only one (Site and Regional) or fewer than five (Local) sequence in the data set were removed prior to analysis. Sequence alignments were run using MUSCLE (align_seqs.py) according to Caporaso et al. (2010b). QIIME was used to calculated all diversity metrics and statistics (alpha_diversity.py and beta_diversity.py) according to Caporaso et al. (2010a). Testing the relationships between OTU distributions and environmental and spatial variables All statistical analyses were performed using QIIME (Caporaso et al., 2010a). For categorical variables, we used Analysis of Similarities (ANOSIM) to determine significance (compare_categories.py). ANOSIM is a non-parametric (permutation-based) test that is similar to Nonmetric Multidimensional Scaling (NMDS) ordination in that it uses the rank order of dissimilarity values (from a distance matrix) across metadata categories (see vegan package in R). Mantel tests were used for numerical variables (compare_distance_matrices.py), and are equivalent to a multivariate Pearson’s correlation. In order to construct the optimal n-parameter model of all available quantitative (numerical) variables, we use BEST analysis (compare_categories.py). BEST will rank variables that explain the largest amount of variance in the data set for a 1-parameter, 2-parameter…n-parameter model (where n is equal to the number of variables provided), and provide a rho-statistic that quantifies the goodness of fit (see ‘bioenv’ in the vegan package for R). Distance-based Redundancy Analysis (db-RDA; constrained ordination) was used in conjunction with forward selection (ordistep function the vegan package for R) to obtain a set of metadata variables that independently (and significantly at P<0.1) explained a portion of the community variance. This subset of non-autocorrelated metadata parameters was used to constrain our Mantel-r comparison in Fig. 1. Procrustes analysis, as implemented in QIIME, was used to compare distributions of samples in PCoA space (Caporaso et al., 2012). References Caporaso JG, Lauber CH, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME Journal 6: 1621-1624. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. 2010a. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7: 335-336. Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010b. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266-267. Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460-2461. Huson DH, Rupp R, Scornavacca C. 2011. Phylogenetic networks: Concepts, algorithms and applications. Cambridge University Press, Cambridge. Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution 30: 772-780 Table S1 Comparisons of Mantel-r and P-values from Mantel tests (based on 1000 permutations on β-diversity distance matrices using Hellinger distances) across the three datasets when OTUs were delineated using the monophyletic clade approach (MCA) or 97% universal threshold. Dataset Variable Site NO3 pH PO4 Soil moisture Spatial coordinate X Spatial coordinate Y Spatial coordinate Y2 Carex arenaria Dianthus deltoides Aspect Elevation pH SOM CEC PO4 K NO3 Mg Ca Spatial coordinates Coarse sand Fine sand Silt Clay Conductivity pH NO3 SOM PO4 S K Mg Ca Na Invasive cover Native cover Spatial coordinates Local Regional MCA Mantel-r P-value 0.079 0.27 0.036 0.48 0.000 0.99 0.073 0.34 -0.039 0.46 0.13 0.010 0.13 0.012 0.17 0.012 0.19 0.023 -0.13 0.24 -0.130 0.38 -0.040 0.74 0.001 0.99 0.011 0.92 0.099 0.30 0.19 0.039 0.11 0.41 -0.081 0.53 -0.068 0.55 0.066 0.44 0.57 0.005 0.38 0.055 0.42 0.032 0.20 0.12 0.17 0.36 0.32 0.025 -0.031 0.85 0.006 0.98 0.25 0.13 0.21 0.014 0.37 0.014 0.32 0.021 0.026 0.91 0.50 0.010 -0.16 0.34 -0.11 0.58 0.55 0.002 97% Mantel-r P-value 0.091 0.24 0.033 0.50 0.020 0.80 0.084 0.38 -0.039 0.47 0.16 0.006 0.16 0.006 0.19 0.014 0.25 0.009 -0.12 0.21 -0.15 0.24 0.019 0.86 0.050 0.59 0.008 0.93 0.09 0.33 0.25 0.008 0.14 0.24 -0.035 0.75 0.003 0.98 0.057 0.46 0.42 0.025 0.33 0.065 0.36 0.045 0.12 0.32 0.26 0.095 0.24 0.054 -0.047 0.76 -0.033 0.81 0.25 0.089 0.12 0.18 0.30 0.029 0.23 0.067 0.023 0.88 0.41 0.023 -0.098 0.52 -0.058 0.73 0.43 0.008 Table S2 Results from ANOSIM and BEST analyses for the three datasets using either the monophyletic clade approach (MCA) or the 97% universal threshold to delineate OTUs. Dataset Analysis Response MCA 97% Site ANOSIMTreatment R-value P-value BEST-rho Parameters -0.0004 0.48 0.12 Soil moisture NO3, pH 0.53 0.0001 0.20 PO4, K 0.068 0.09 0.67 0.001 0.63 0.001 0.59 S, silt, coarse sand -0.0149 0.61 0.14 Soil moisture NO3, pH 0.62 0.0001 0.27 PO4, K 0.15 0.029 0.49 0.008 0.43 0.003 0.49 pH, clay, silt, coarse sand BEST Local ANOSIMCover type BEST Regional ANOSIMPlant species ANOSIMRegion ANOSIMSite BEST R-value P-value BEST-rho Parameters R-value P-value R-value P-value R-value P-value BEST-rho Parameters Table S3 Correlations of the beta-diversity distance matrices (Hellinger distances) and Procrustes analyses of MCA versus 97% OTU delineation approach for the three datasets. Dataset Analysis Parameter Value Site Correlation Mantel-r P-value M2 P-value Mantel-r P-value M2 P-value Mantel-r P-value M2 P-value 0.98 0.001 0.000 0.008 0.94 0.001 0.089 <0.0001 0.77 0.001 0.42 <0.001 Procrustes analysis Local Correlation Procrustes analysis Regional Correlation Procrustes analysis Supporting Information Figs S1–S5 Fig. S1 NeighborNet split network on the Site dataset based on the MCA approach that identified 33 OTUs and a 97% universal threshold that identified 76 OTUs (b). Fig. S2 NeighborNet split network based on the MCA approach that identified 46 OTUs (a, as in Lekberg et al. 2013) and a 97% universal threshold that identified 1083 OTUs (b). Fig. S3 NeighborNet split network on the Regional data set based on the MCA approach that identified 30 OTUs (a) and a 97% universal threshold that identified 278 OTUs (b). Fig. S4 Procrustes analysis of AMF community samples for the Site (a) and Regional (b) datasets. Duplicate points represent data processed through the MCA and 97% OTU pipelines. The lines between points highlight the Euclidean distance between points that represent the two different methods. Fig. S5 Supervised learning results for the Local dataset, showing an exponential increase in OTU numbers and varying classification error over a 90–99% OTU threshold range. The error indicates the likelihood of assigning an unknown AMF community to the incorrect aboveground plant communities (cheatgrass, knapweed, leafy spurge, and native) after training the learning algorithm on a subset of the data. Error bars for the classification errors were determined via 10-fold cross validation. The 98% threshold differed from the 95% (F = 4.77, P = 0.043) and 97% (F = 5.09, P = 0.037), but these differences disappeared after controlling for multiple comparisons. Fig. S1 Fig. S2 Fig. S3 Fig. S4 Fig. S5