Liver Diseases in Childhood
advertisement

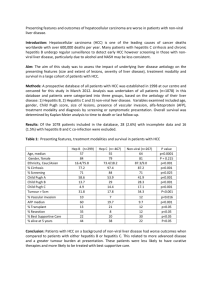
Liver Diseases in Children by Dr. AlSarkhy Introduction Main cholesterol is from the body. Synthetic liver function s affected when the liver fails. o Ammonia is toxic to the liver and urea and is not. Patients who are cirrhotic have no cells to release AST, ALT). Patients with Hepatitis B, C may have normal ALT, AST; therefore, LFTs do not reflect the severity of the disease. GGT is a more specific liver marker in pediatric patients, in comparison to ALP in adults. In the pediatric age group, ALP will be high in the growing child and also during pregnancy (this is not necessarily true in adults). Pale stool/ abnormal stool should always be taken seriously. o In children, most commonly biliary atresia or any sort of obstruction. o In adults, think of malignancy. o Abnormal stool can be pale, red, or black. In all these forms, full investigations should be carried out. Main and commonest presentation of liver disease is jaundice. Any child who presents with jaundice after 2 weeks of birth is pathological until proven otherwise (except breast milk jaundice that can extend up to 1 month). Jaundice Usually the picture is not completely choleostatic or hepatitic, it can present with a mixture of both. Cholestasis (conjugated) or direct: can be mechanical or functional i.e. problem with receptor or enzymatic problem. o When bile acid deposits in the skin it causes itching whereas cholesterol causes xanthoma mostly around the eye, upper limb, lower limb and tendon sheaths. Xanthomas in pediatric age group do not increase the cardiovascular risks. Biliary atresia Worldwide, biliary atresia is the most common cause of liver transplant. In KSA, due to the high incidence of consanguineous marriages, metabolic diseases are the most common cause of liver transplantation. Biliary atresia is mostly extrahepatic. Biliary atresia can start before delivery or after. Thought to be either autoimmune or inflammatory. o Mother gets viral infection during pregnancy, leading to antibodies attacking the biliary tract, which may ascend to liver leading to cirrhosis and end stage liver disease and death. Therefore, urgent diagnosis in mandatory. Typically presents after 2 weeks with acholic (pale) stool. If it is not treated before two years, patient may be cirrhotic and die. Diagnosis o Abdominal US to visualize the obstruction, not diagnose atresia. 1 o HIDA scan: child will take up dye, and if he has biliary atresia, the hepatocyte will take the dye but not excrete it. o Confirmatory test: liver biopsy. o Confirmatory test and definitive diagnosis is by intra-op cholangiogram because you will see that when dye is injected in to the gall bladder it will go to the liver. Treatment: surgery should be done before the age of 2 by connecting the porta hepatis (where bile is excreted) to the jejunum. o If surgery was done before 2 mnths, there’s a 60% chance of not needing transplant. o If surgery failed or done later in life (> 2 months), the operation has a higher risk of failure and need for liver transplantation later in life. Choledocal cyst Abnormal dilatation of biliary system. Can be asymptomatic or may present clinically similar to biliary atresia and can cause obstruction to biliary tree or small bowel. Therefore, biliary atresia is an important differential diagnosis. o When totally asymptomatic, can be discovered by autopsy. Cholagocarcinoma is a risk later in life. Must be resected once discovered. Alpha 1 anti-trypsin deficiency Alpha anti-trypsin is a protease inhibitor that inhibits elastase destruction. Autosomal recessive disease. Normally, the alpha 1 is formed from Pi MM gene. If Pi ZZ, then it’s abnormal and acquire the disease. While the carrier state is Pi MZ. In adults, presentation is mainly pulmonary while it’s hepatic in pediatric patients. Alpha 1 anti-trypsin formed in the liver (abnormal) that is not excreted and accumulats in the liver leading to inflammation and cirrhosis. There is no cure, unless it reaches end stage liver disease which requires liver transplantation. Some patients do not require any treatment. Neonatal Hepatitis Previously, when all causes of hepatitis have been excluded, it would be classified as neonatal hepatitis as an idiopathic cause. However, due to the discovery of multiple causes of hepatitis, neonatal hepatitis is becoming a rare entity. Liver disease in older children is similar to adults Acute hepatitis Hepatitis A It is the most common type. Simple (usually without jaundice but may sometimes present with jaundice) and goes without any problems. Very rarely progresses to fulminant hepatitis and require liver transplantation. In adults, on the other hand it is a severe disease. 2 It is usually part of an endemic. No chronic carrier state, and its diagnosed by anti-IgM. Treatment is supportive. Hepatitis A is a reportable disease that must be reported to the ministry of health. Vaccine of hepatitis A (2 doses at 18 and 24 months) can also be given to adults. Hepatitis B Can present as both acute and chronic. Transmission is mainly vertical, and also due to blood transfusion seen in patients with thalassemia or sicklers. Majority of pediatric patients with hepatitis B go to chronic state because of their poor immunity at birth. Therefore, the earlier they acquire the disease, the worse the prognosis. Whereas in adults the disease is mostly cleared by their good immunity. Serology o HBcAb tells you that the virus was here. IgG is from vaccine or previous vaccine, whereas IgM indicates acute infection. o HBsAg and HBsAb never co-exist if one is there the other disappears, except during the window period where both the Ag and Ab are absent which is period of disappearance of the antigen and appearance of the antibodies. Treatment o If the mother during pregnancy is found to be HBsAg positive then the baby is treated immediately with both immunoglobulin within 12 hours of birth, and vaccined within 7 days. This is 95% effective. o There are usually two hits for the virus one by targeting immunity and other by the drug. If the child is found to have the disease at birth and its not symptomatic then we treat with immunoglobulins and vaccine. If later in life they become symptomatic, then drug therapy is the preferred method of choice. o In children, usually the disease is not active at first so we wait and watch the LFTs. Medication are not effective and have a lot of side effects. The virus is not the problem; it is the immunity itself that destructs the hepatocytes. Therefore, we usually wait and see unless it progress to chronic liver disease then we start medications. Hepatitis C Positive antibodies on PCR or blood are diagnostic. Treatment with medication because there’s no vaccine and it’s more destructive thus symptoms outweigh side effects. Therefore, more eager to treat. Hepatitis D It’s seen in drug abusers. Hepatitis E Very bad during pregnancy (miscarriage, abortions). 3 Autoimmune Hepatitis Very common in adolescents > 9 years old. Autoantbodies from viral infection fight the virus and hepatocytes. Can progress to CLD then to liver transplantation. Diagnosis is by more than 1 parameter o Elevated ALT and AST more than GGT and ALP (hepatatic picture). o Elevated immunoglobulins. o Liver disease signs. Treatment by steroid and azathioprine, and carries a good progosis. Wilson disease It is and autosomal recessive disease. If you see one patient with the disease, screen the family. Pathophysiology: the copper cannot be excreted from the biliary tree due to genetic defect. The copper can also be deposited in the brain (thalamus). Presentation: typically presents with liver disease but also can present with mental illness and the illness is treated by treating the Wilson. o Can also present with fulminant hepatic failure due to toxic accumulation of copper. o It is usually a coombs –ve hemolytic crisis (Coombs positive is usually seen with autoimmune hemolytic anemia and is treated with steroids). Diagnosis o 24 hours urine collection of Copper, wheich is usually enough. o Liver biopsy. o Genetic testing, but it takes 2-3 months time. o Any patient with liver disease under the age 4 should have work-up done for Wilson’s disease. Treatment is by chelating agent “penicillamine” and Zinc. Crigglar Najjar syndrome If a patient presents with unconjugated hyper-bilirubinemia and normal LFTs, it’s Crigglar Najjar. If not treated, it can progress to kernicterus. Types o Type 1 is treated with phototherapy for 20 hours a day for the rest of their life. Might require transfusion early in life and might go for liver transplant. o Type 2 is treated with phenobarbitone. Gilbert syndrome Very benign familial condition. Treated with hydration and reassurance. Expect the condition to get worse with infection. Ischemic hepatitis ALT/AST in thousands. If you correct volume, LFT will go back to normal. 4