Multiscale Modeling Study of Graphene Oxides
advertisement

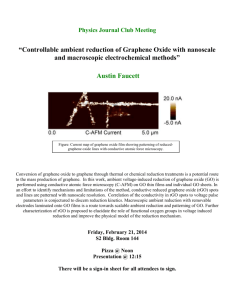
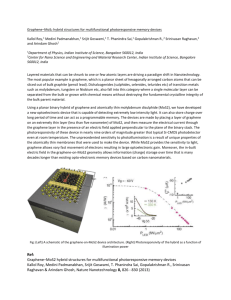
“Multiscale Modeling Study of Graphene Oxides” Kyeongjae (KJ) Cho, UT Dallas Thursday, Feb. 17, 2011 10:45 a.m., NSERL 3.204 Abstract Graphene has been seriously considered for nanoelectronic devices because of its high electrical mobility, high thermal conductivity and other promising material properties. In any practical electronic device systems, electron transporting materials (silicon or graphene) need to be controlled by insulating materials (SiO2 or graphene oxide), which can function as gate dielectrics or a separator between device structures. Thus the role of graphene oxide (GO) in graphene-based nanoelectronics is comparable to that of SiO2 in silicon-based microelectronics. Consequently the graphene electronic research community is investigating diverse oxides (e.g., GO and alumina grown on graphene using ALD) as candidates for gate oxides. However, the atomic and electronic structures of GO and their impact on electrical properties have not been fully understood. In this talk we will discuss the quantitative nature of the chemical and physical properties of GO based on atomistic and quantum simulations in close collaboration with experimental groups. Our early modeling study has shown that graphene basal plane is relative inert. Nevertheless, ozone was suggested to activate the basal plane for subsequent chemical reactions, and a detailed modeling study has confirmed the epoxide forming reaction of ozone treatment. Basal plane oxide of graphene is known to etch carbon atoms by CO and CO2 gas species, and etch holes are formed in the carbon planes of GO structures. The graphene edge oxide species are predicted to have metallic properties, and recent IR experiments have demonstrated the presence of free electrons at the oxide edges of reduced GO. Furthermore, edge oxide structures are shown to be formed by interactions with water molecules during the thermal annealing of the GO. These studies are providing important clues on the nature of GO. Bio KJ Cho is an associate professor of materials science and engineering and of physics. Before joining UT Dallas in 2006, he was an assistant professor of mechanical engineering and of materials science and engineering at Stanford University. His research interests concern computational modeling of nanomaterials with applications to nanoelectronic devices and renewable energy technology. For materials modeling, his research group has developed an atomistic modeling method to simulate atomic structures of nanomaterials and a tight binding method to calculate electronic structures and quantum transport properties of nanoelectronic devices. Advanced first principles quantum simulations methods (density functional theory) are used to investigate the nanomaterials with quantitative accuracy and achieve fundamental understanding of structure-property relationships. # # #