Electronic Supplementary Material Methods (a) Sequence assembly
advertisement
 Sequence assembly")
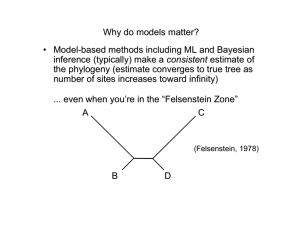
Electronic Supplementary Material Methods (a) Sequence assembly and orthology assignment Sequenced results were quality filtered accordingly to a threshold average quality Phred score of 30 and adaptors trimmed using Trimgalore v 0.3.3 [1]. Ribosomal RNA (rRNA) was filtered out using Bowtie v.1.0.0 [2]. De novo strand-specific assemblies were made using Trinity [3,4] using paired read files, with an enforced path reinforcement distance of 75. Resulting assemblies were processed in TransDecoder [4] in order to identify candidate open reading frames (ORFs) within the transcripts. Redundancy reduction was done with CD-HIT [5] for all Trinity assemblies (95% similarity). Predicted peptides were then processed with a further filter to select only one peptide per putative unigene, by choosing the longest ORF per Trinity subcomponent, thus removing variation in coding regions attributable to alternative splicing, closely-related paralogs, and allelic diversity. Predicted ORFs were assigned to orthologous groups using the Orthologous MAtrix (OMA) algorithm, (OMA stand-alone v.0.99u; [6,7]), which has been shown to outperform alternative approaches (e.g., reciprocal best hit; OrthoDB) toward identification of true orthologs and to minimize Type I error in orthology assignment [8]. Additional taxa not sequenced by us (table S1) were obtained from GenBank and processed in TransDecoder. For Sanger-sequenced EST and 454 libraries, redundancy reduction was done with CD-HIT as described above. Due to the small size of additional data sets and/or the quality of the genome of Mesobuthus martensii, predicted ORFs were assigned to orthologous groups using OMA in two separate runs, one for Buthidae (Iurus, Chaerilus, and the pseudochactids used as outgroups; taxon occupancy criterion set to representation in at least 19 taxa) and a second for “Chactoidea” and Scorpionoidea sensu stricto (Iurus and Bothriurus used as outgroups; taxon occupancy criterion set to representation in at least 16 taxa). This was done for computational expediency as well as to ensure representation of the smallest libraries in supermatrices. (b) Heuristics in phylogenomic analysis Maximum likelihood (ML) analyses were conducted using RAxML v.7.7.5 [9] and Bayesian inference (BI) using PhyloBayes MPI 1.4e [10]. For RAxML v.7.7.5, we implemented a unique CAT + LG4X + F model for each gene [11], with 100 independent starts for each analysis. Bootstrap resampling frequencies were estimated with 250-500 replicates using a rapid bootstrapping algorithm [12]. Analyses with PhyloBayes MPI 1.4e were limited to smaller datasets (Matrices 1-2), as implementation of PhyloBayes for large matrices requires intractable amounts of time for convergence. We implemented PhyloBayes MPI 1.4e using the siteheterogeneous CAT + GTR model of evolution [13]. Four independent chains were run for 10,216-59,229 cycles, depending on the data set analyzed. The initial 5,000 cycles were discarded as burnin, with convergence assessed using the maximum bipartition discrepancies across chains. For both matrices, the chains did not converge (maximum bipartition discrepancies approximately equal to 0.7), but ingroup topologies were identical and well supported (posterior probability ≥ 0.97); failure to converge was driven by uncertainty in the relative placement of two outgroup terminals (alternating positions of Eremobates sp. and Ricinoides atewa as sister group to Opiliones). We incorporated a branch length mixture model of heterotachy, a model that has been shown to outperform covarion and homotachous counterparts when heterotachy is incident [14]. A maximum likelihood approach was implemented using the PhyML v.3 extension software M3L [14-16]. We analyzed our two most complete matrices (Matrix 1 and Matrix 11; both >90% occupancy) using a branch length mixtures model (+B) with four mixture categories in conjunction with the LG substitution model. A best-of-SPR-and-NNI branch-swapping algorithm was selected to optimize tree topology, starting with a BioNJ tree. For both analyses, a single mixture was selected (weight: 1), and all other mixture classes received a weight of 0. Basal scorpion relationships in both analyses were consistent with their CAT-GTR and/or CAT+LG4X counterpart topologies. (c) Statistical tests of non-random distribution of missing data To infer the impact of non-randomly distributed missing data, we treated the absence or presence of genes as cells in a matrix and quantified the degree of phylogenetic structure in the unoccupied cells for each of the 5,025 genes. Using custom R and Python scripts, we scored the parsimony length of each column (gene) on the tree topology shown in figure 2c. For a given gene, random distribution of missing data is expected to yield a high parsimony score, whereas non-random (clade-specific) distribution of missing data is expected to yield a low parsimony score. We then compared this to a null distribution of parsimony scores obtained for the corresponding column upon randomly reshuffling terminals 1,000 times. If a column’s parsimony length was below the lower bound of the 95% confidence interval of parsimony scores in the null distribution, we scored the orthogroup as having significantly non-randomly distributed missing data. This strategy is a modification of an older approach (cladistic permutation tail probability or PTP test; [17]), but inverts the interpretation of the result. As applied here, a significant result means that the distribution of a gene’s absences in the matrix is non-random and should therefore be discarded from analysis. As the original PTP test was shown to have a high type I error, the test we apply is highly conservative. (d) Bayesian relative rates test To assess the possibility of long-branch artifacts affecting the ingroup, a Bayesian relative rates test (sensu [18]) was conducted as follows: from the Bayesian inference analysis of Matrix 2, 500 post-burnin trees from each of the four PhyloBayes chains were randomly selected. All 2,000 trees were rooted with Limulus polyphemus, and all chelicerate orders represented by a single terminal were culled (Limulus polyphemus, Eremobates sp., and Ricinoides atewa). The patristic distances from the root to each terminal was calculated across the set of 2,000 trees. The resulting set of distances for each terminal was used to conduct statistical comparison of branch length distributions [18]. References 1. Wu ZP, Wang X, Zhang XG. 2011. Using non-uniform read distribution models to improve isoform expression inference in RNA-Seq. Bioinformatics 27:502-508. 2. Langmead B, Trapnell C, Pop M, Salzberg SL. 2009 Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25. 3. Grabherr MG et al. 2011 Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnology 29, 644-652. (doi:10. 1038/nbt.1883) 4. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. 2013 De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494-1512. 5. Fu LM, Niu BF, Zhu ZW, Wu ST, Li WZ. 2012 CD-HIT: accelerated for clustering the nextgeneration sequencing data. Bioinformatics 28, 3150-3152. 6. Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. 2011 OMA 2011: Orthology inference among 1000 complete genomes. Nucleic Acids Research 39, D289-D294. 7. Altenhoff AM, Gil M, Gonnet GH, Dessimoz C. 2013 Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS ONE 8, e53786. 8. Altenhoff AM, Dessimoz C. 2009 Phylogenetic and Functional Assessment of Orthologs Inference Projects and Methods. PLoS Computational Biology 5, e1000262. 9. Berger SA, Krompass D, Stamatakis A. 2011 Performance, accuracy, and Web server for evolutionary placement of short sequence reads under maximum likelihood. Systematic Biology 60, 291-302. 10. Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013 PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Systematic Biology 62, 611-615. 11. Le SQ, Dang CC, Gascuel O. 2012 Modeling protein evolution with several amino acid replacement matrices depending on site rates. Molecular Biology and Evolution 29, 2921-2936. 12. Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Systematic Biology 57, 758-771. 13. Lartillot N, Philippe H. 2004 A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Molecular Biology and Evolution 21, 1095-1109. 14. Kolaczkowski B, Thornton JW. 2008 A Mixed Branch Length Model of Heterotachy Improves Phylogenetic Accuracy. Molecular Biology and Evolution 25, 1054-1066. 15. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010 New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59, 307-21. 16. Hanson-Smith V. 2013 M3L. Google Code Repository (https://code.google.com/p/m3l/) 17. Faith DP. 1991. Cladistic permutation tests for monophyly and nonmonophyly. Systematic Zoology 40, 366-375. 18. Wilcox TP, García de Léon FJ, Hendrickson DA, Hillis DM. 2004. Convergence among cave catfishes: long-branch attraction and a Bayesian relative rates test. Molecular Phylogenetics and Evolution 31, 1101-1103.