Nawijn Abstract
advertisement

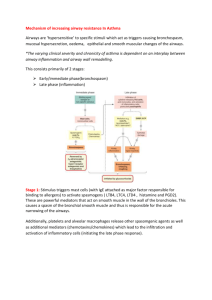
Asthma as a disease of the airway epithelium: Protocadherin-1 and E-cadherine in allergic airway inflammation and hyperreactivity Martijn C. Nawijn1,2 (1) University of Groningen, University Medical Center Groningen, Department of Pathology & Medical Biology, laboratory of Experimental Pulmonology and Inflammation Research (EXPIRE), Groningen, The Netherlands (2) University of Groningen, University Medical Center Groningen, GRIAC Research Institute, Groningen, the Netherlands Abstract The airway epithelium in altered in asthma, with a reduced expression of tight and adherens junctions proteins, increased permeability to small molecules and increased release of proinflammatory mediators in vitro. The identification of a large number of airway-epithelially expressed genes as asthma genes in genetic and genomic analyses indicates a possible contribution of the airway epithelium to the inception of asthma. It is, however, currently unknown whether the impaired barrier function and reduced expression of adherens and tight junction proteins in the airway epithelium of asthmatic patients is a cause or a consequence of the disease. Here, we show that loss of the adherens junction molecule E-cadherin or the human asthma and BHR gene Protocadherin-1 directly affects the barrier function and inflammatory phenotype of the airway epithelium and induces spontaneous phenotypes remiscent of asthma as well as an increased susceptibility to allergens in vivo. Mice with conditional loss of Ecadherin in the airway epithelium (Cdh1fl/fl Cre+) were compared to Cre-negative littermate controls (Cdh1fl/fl Cre-). Naïve mice were examined for lung anatomy and inflammation on day of birth and at 2 weeks (2W), 4W, 6W and 8W of age, and after House Dust Mite treatment (HDM) in 4W old mice. Mice deficient for the human asthma gene Pcdh1 were studies at baseline and after HDM challenges at 8 weeks of age in comparison to wild-type littermates. Cdh1fl/fl Cre+ mice were viable and displayed no gross abnormalities in lung development. However, pro-inflammatory activation of the airway epithelium was evident from CCL17 and CCL20 production, and inflammatory dendritic cells and eosinophil accumulated in lungs of naïve Cdh1fl/fl Cre+ mice from W2 onwards. Cdh1fl/fl Cre+ mice display spontaneous and progressive hypoplasia of ciliated and secretory airway epithelial cells in the bronchioles from birth onwards, associated with spontaneous goblet cell metaplasia at later ages. Exposure of Cdh1fl/fl Cre+ mice to HDM at 4W of age induced strongly exaggerated eosinophilic airway inflammation in compared to Cre- littermates controls. The human asthma gene PCDH1 was found to encode an adhesion molecule of the cadherin superfamily. PCDH1 was found to contribute to airway epithelial barrier function at baseline as well as during wound repair. Loss of PCDH1 in vivo resulted in a airway hyperresponsiveness at baseline as well as an exaggerated inflammatory response to HDM exposure. These data show that loss of airway epithelial integrity is not merely a consequence of asthma, but can in itself cause airway structural and inflammatory phenotypes reminiscent of asthma. The validation of these phenotypes in mice lacking a human asthma gene that contributes to airway epithelial barrier function underscores the relevance of these translational studies for human disease, identifying airway epithelial integrity as a target for novel therapeutic interventions.