Admixture14
advertisement

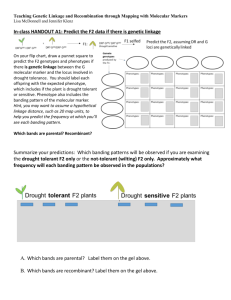
Abdul Ahad Sercan Kobazoglu Mohammed El-kholy Tugrul Bardak Nature Reviews – Mapping By Admixture Linkage Disequilibrium: Advances, Limitations and Guidelines (Michael W. Smith & Stephen J. O’Brien) Principles Illustrated In Paper Linkage disequilibrium is the principle where members of a certain population show a non-random proportion of linked genes. Mapping admixture linkage disequilibrium (MALD) is a useful technique in mapping and identifying short nucleotide polymorphisms (SNPs) that can be indicators of disease. It uses the principle of linkage disequilibrium to look at an admixed population (a population that is created through interbreeding of two previously separated populations), thus it makes use of the resultant haplotypes produced by gene flow as a result of admixture that has taken place between these two populations which use to be separate. Furthermore, the papers look at the non-random association of alleles at different loci. Linkage disequilibrium can be caused by the genes physically being linked on a chromosome due to selection. MLAD can be used to map complex genetic diseases via indirect association. Mutations at loci close to the potential ‘disease causing gene’ are mapped and can be indicators of human diseases. Using these SNP's rare Mendelian diseases can be identified. Mapping admixture linkage disequilibrium is useful for studying the haplotypes of the recent admixed African-American populations. Alternative Hypothesis In this paper there is no specific hypothesis studied per se. It does however; suggest that methods alternative to MLAD are available and should be explored, these being: linkage mapping and genetic association mapping. Therefore, this paper discusses mapping by admixture linkage disequilibrium and genetic association mapping. It weights up the advantages and disadvantages of both techniques. Linkage mapping is only useful for identifying rare monogenic Mendelian diseases. Genetic association mapping on the other hand is a much more powerful tool, at identifying complex multifactorial disease. However, it requires many more markers and is considerably more expensive. The paper then goes on to explain, that using MALD is a more preferable technique as it is economically more feasible and includes around 2000 markers only, additionally, it will be more reliable as the accuracy when studying the effects of multiple genes on a complex disease is higher. Data Collected This study uses data which has been collected using genomic data from ethic groups around the world; it applied the MALD technique to those of African, American and European decent. What makes this study interesting is that only genomes of individuals who have mixed ancestry and suffer from certain diseases of interst to researchers were selected. Generating appropriate sample sizes required to make a reliable experiment in order to estimate the number of disease related alleles, varied depending on parental ancestries and the disease that was being investigated. Preliminary results indicated that African-American populations had an increased likelihood of carrying alleles that were associated with complex genetic diseases such as, Type II Diabetes. If the assumptions of this study are correct, this would in turn mean that African parental populations contributed to the pool of disease associated alleles among the generation in question. Although, one must proceed with caution when analysing and interpreting these results, as they are preliminary and are only estimates based on sample sizes – thus no result is conclusive. Abdul Ahad Sercan Kobazoglu Mohammed El-kholy Tugrul Bardak Data interpretation Currently, MALD is in its early stages, it is hoped that in the future it will provide costefficient and accurate way of determining high risk disease associated alleles in order to help prevent and treat diseases that have a genetic predisposition. The genomic data collected from mixed ethnic groups that have diseases of interest were screened against a vast set of polymorphic markers, which allowed for ancestral identification. Furthermore, regions of genome that are known to have a considerable number of genes that are associated with diseases were also integrated into the study, which was achieved by identifying short nucleotide polymorphisms (SNPs) and combining that data with linkage mapping to map the genome and certain alleles that may influence certain diseases. Once a statistic has been calculated to determine the likelihood of an individual developing a disease, it is put against criteria to determine whether the result can be declared as being significant; this includes applying a Bayesian approach (Box 1). Box 1: Criteria for declaring significance in a MALD study Figure 1: Detecting disease associated genomic regions using mapping by admixture linkage disequilibrium.