Supplementary Text (doc 34K)
advertisement
")
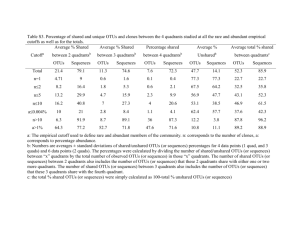
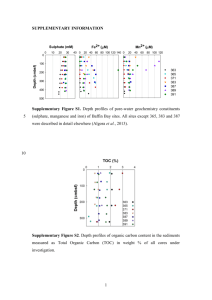
Supplementary text Overview of sequencing statistics. The V4-V5 regions of the 16S rRNA genes were amplified using a conserved primer pair, which was tagged with a short oligonucleotide sequence of 6 bp (i.e. barcodes) (Table S1). These samples were sequenced with one pyrosequencing run, and a total of 52.2 mbp sequences with 213,329 reads were obtained. Since both forward and reverse primers were tagged, sequences from both strands were obtained. Although PCR amplification with this primer set generated ~400 bp fragments, only the first 240 bp after the proximal PCR primer was used for analysis due to sequence quality problem. The sequences from both forward and reverse primers were analyzed separately or combined together. A total of 178,148 raw sequencing reads were obtained, ranging from 1,637 to 8,275 sequences per sample prior to sequence preprocessing (Table S7-C). After removing low quality reads as described in the Materials and Methods section, a total of 115,741 sequence reads were obtained (24 samples, 56 tags). About 6% (p=0.049 based on Mann-Whitney U test) more sequence reads were obtained with the forward primer (52.8%) than reverse primer (47.2%). The number of total pyrosequencing reads varied considerably among different tags, ranging from 432 to 2,999 with an average of 1,091 reads for the forward primers (Table S7-A), and from 487 to 3,158 with an average of 976 reads for the reverse primers (Table S7-B). These results are similar to other pyrosequencing results (Campbell et al., 2010; Hollister et al., 2010; Acosta-Martinez et al., 2008; Koopman et al., 2010; Middelbos et al., 2010). All pyrosequencing reads from all samples were aligned using the RDP Infernal Aligner and the complete linkage clustering method was used to define OTUs using 97% sequence identity as a cutoff. A total of 17,218 OTUs were obtained. About 17% (p<0.001) more OTUs were obtained with the forward primer (58.6%) than reverse (41.4%). The numbers of OTUs from different tags vary significantly, ranging from 306 to 1,480, with an average of 633 OTUs per tag, for the forward primer (Table S7-A), and 235 to 1,358, with an average of 502 OTUs per tag, for the reverse primer (Table S7-B). Rarefaction analysis of microbial community diversity. To understand whether the OTUs obtained via pyrosequencing represent the diversity of the abundant populations of microbial communities, rarefaction analysis was carried out based on individual primers, combined primers, individual plots, treatments and the experimental site as a whole. The majority (91.1%) of the experimental data fitted well with a nonlinear exponential model, y = ax (1 – exp (-b*x) (Zhou et al., 2004) (r2 = 0.9727 - 0.9999). Most of the rarefaction curves (plots of the cumulative number of OTUs as a function of sequence number) approached saturation (Fig. S3 and data not shown), suggesting that sampling efforts were reasonably sufficient to reflect the diversity of the communities examined. Based on the rarefaction curves, predicted OTU numbers for each sequencing tag were obtained. Overall, about 57±7.2% of the predicted OTUs were sampled with different tags for the forward primers (Table S7-A), 64±6.0% for reverse primers (Table S7-B) and 51±6.4% for both combined (Table S7-C, p values: F/R, <0.001; F/FR, <0.001; R/FR, <0.001). Also, based on Chao1 estimation, about 37±4.5% of the predicted OTUs were sampled with different tags for forward primers (Table S7-A), 41±4.9% for reverse primers (Table S7-B) and 32.6±4.2% for both combined (Table S7-C, p values: F/R, <0.001; F/FR, <0.001; R/FR, <0.001). In addition, a total of 16,825 OTUs were observed by pooling all sequences together, which was 96.8% of the OTUs predicted by rarefaction analysis (17,370), and 85.2% of the OTUs based on Chao1 (19,746) (Table S7-C), indicating that the diversity of the abundant populations in these communities was recovered in this study. Key issues for improving data comparability. To make a meaningful biological comparison across different experimental conditions from noisy data, the simplest way to improve the data comparability is to increase sequencing efforts and generate sufficient number of sequences from each sample. Nevertheless, this can be difficult due to the complexity of soil microbial communities and experimental cost. Another improvement is to remove less confident sequence data, such as singleton sequences and/or less representative OTUs (e.g., OTUs present only once or twice across samples) (He et al., 2010). In addition, increasing biological replicates should also be an effective improvement. In the following, we systematically examined the effects of these various strategies on improving data comparability using the data from this experimental site. Previous studies demonstrated that experimental warming at this site markedly affected plant growth and phenology (Luo et al. 2007; Sherry et al., 2008), as well as microbial community structure based on fatty acid analysis (Zhang et al., 2005) and functional gene arrays (Zhou et al, unpublished). The aboveground community structures also differ between clipping and non-clipping. It is thus reasonable to believe that warming or clipping will significantly shift microbial community phylogenetic composition and structure. Therefore, we assessed the effectiveness of various data preprocessing strategies for improving data comparability by simply judging whether they can provide better differentiation (resolution) of microbial community phylogenetic structure between warming and unwarming, or between clipping and unclipping. In this study, some of the samples (plots) were sequenced with 2 tags and the others with 3 tags. This data can be combined in two ways to represent the community composition and structure of a sample. One is to add all OTUs together by treating the sequencing with these tags as a continuous sampling event, which leads to increased sequencing effort. The alternative way is to calculate the average numbers of OTUs identified with these tags by considering the sequencing with different tags as replicate sampling events. The latter could lead to increased sequencing efforts, but to lesser extent. Thus, the former approach is generally preferred. In addition, since the sequence number of individual OTUs obtained varied significantly among different samples, the relative proportions of sequence numbers were used for community comparison. The main results based on the above considerations were described in the main text. References 1. Acosta-Martinez V, Dowd S, Sun Y, Allen V (2008). Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol Biochem 40: 2762-2770. 2. Chao A (1987). Estimating the population-size for capture recapture data with unequal catchability. Biometrics 43: 783-791. 3. Campbell BJ, Polson SW, Hanson TE, Mack MC, Schuur EAG (2010). The effect of nutrient deposition on bacterial communities in Arctic tundra soil. Environ Microbiol 12: 1842-1854. 4. Koopman MM, Fuselier DM, Hird S, Carstens BC (2010). The Carnivorous Pale Pitcher Plant Harbors Diverse, Distinct, and Time-Dependent Bacterial Communities. Appl Environ Microbiol 76: 1851-1860. 5. He Z, Xu MY, Deng Y, Kang SH, Kellogg L, Wu LY et al (2010). Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO2. Ecol Lett 13: 564-575. 6. Hollister EB, Engledow AS, Hammett AJM, Provin TL, Wilkinson HH, Gentry TJ (2010). Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4: 829-838. 7. Luo YQ (2007). Terrestrial carbon-cycle feedback to climate warming. Annu Rev Ecol Evol Syst 38: 683-712. 8. Middelbos IS, Boler BMV, Qu A, White BA, Swanson KS, Fahey GC (2010). Phylogenetic Characterization of Fecal Microbial Communities of Dogs Fed Diets with or without Supplemental Dietary Fiber Using 454 Pyrosequencing. PloS One 5. 9. Sherry RA, Zhou XH, Gu SL, Arnone JA, Schimel DS, Verburg PS et al (2007). Divergence of reproductive phenology under climate warming. Natl Acad Sci U S A 104: 198-202. 10. Zhang W, Parker KM, Luo Y, Wan S, Wallace LL, Hu S (2005). Soil microbial responses to experimental warming and clipping in a tallgrass prairie. Global Change Biol 11: 266-277. 11. Zhou JZ, Xia BC, Huang H, Palumbo AV, Tiedje JM (2004). Microbial diversity and heterogeneity in sandy subsurface soils. Appl Environ Microbiol 70: 1723-1734.