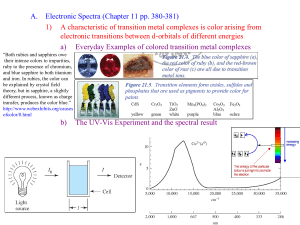
msc_pre_chemistry_pap1_bl2
advertisement

M.Sc. (Previous) Chemistry Paper – I : INORGANIC CHEMISTRY BLOCK – II UNIT – 4 : Magnetic properties of Transition Metal UNIT – 5 : Metal π Complexes UNIT – 6 : Reaction Mechanism of Transition Metal Author – Dr. Purushottam B. Chakrawarti Edtor – Dr. M.P. Agnihotri UNIT-4 MAGNETIC PROPERTIES OF TRANSITION METAL COMPLEXE Structure 4.0 Introduction 4.1 Objectives 4.2 Magnetic Moments 4.3 4.2.1 Number of Unpaired Electrons 4.2.2 Spin Only Formula Anomalous Magnetic Moments 4.3.1 Orbital Contribution in Magnetic Moments 4.3.2 Curie's Law 4.4 Magnetic Exchange Coupling 4.5 Let Us Sum Up 4.6 Check Your Progress: The Key 4.0 INTRODUCTION Substances were first classified as diamagnetic or paramagnetic by M.Faraday (1845). But it was not untill many years later that these phenomenon came to be understand in terms of electronic structures. When any substance is placed in an external magnetic field, there is an induced circulation of electrons producing a net magnetic moment aligned in opposition to the applied field. This is the diamagnetic effect and it arises from paired electrons within a sample. Paramagnetism is produced by unpaired electrons in a sample. The spin and Orbital motion of these electrons give rise to permanent molecular moments that tend to alignt themselves with an applied field. Magnetic properties and electronic spectra are closely connected. Magnetic susceptibility measurements are used to decide between different electronic configurations. It may be mentioned, although the electronic spectra is a powerful method for investigating transition metal complexes, additional and complementary information can be provided by magnetic measurements. In this unit we shall discuss how net magnetic moments of transition metal complexes can be worked out; and in what conditions anomalous magnetic moments are obtained. However, it will be advantageous if you recall what you have already studied earlier about the basic concepts of magnetic moments of atoms. 4.1 OBJECTIVES The main aim of this unit is to study magnetic properties of transition metal complexes and to establish their correlation with their spectral properties. After going through this unit you should be able to: calculate magnetic moments and number of unpaired electrons in a transition metal complex; describe under what conditions spin-only formula will be useful to calculate µ of the complexes; discuss under which conditions orbital contributions will be important to calculate µ of the complexes; and explain magnetic exchange coupling and spin crossover to describe anomalous magnetic moments of some complexes. 4.2 MAGNTIC MOMENTS When a substance is subjected to a magnetic field, H, a magnetization, I, is induced. The ratio I/H is called the "volume susceptibility", K, and can be measured by a variety of techniques, including the Gouy balance method, the Faraday method, and an nmr method. The volume susceptibility is simply related to the "gram susceptibility," x, and the "molar susceptibility", xm x K d xM K M d where d and M are the density and molecular weight of the substance, respectively. For most substances, K, x and xM have negative values; such substances are weakly repelled by a magnetic field and are called "diamagnetic". For substances having unpaired electrons that do not strongly interact with one another, K, x and xM have relatively large positive values; these substances are attracted into a magnetic field and are called 'paramagnetic." When a paramagnetic substance is placed in a magnetic field, the moments of the paramagnetic molecules or ions tend to align with the field; however, thermal agitation tends to randomize the orientations of the individual moments. Theoretical analysis of the situation leads to the relations; x Mcorr N 2 3kT where X Mcorr is the molar susceptibility which has been corrected both for the diamagnetic contribution to the susceptibility (due to the non paramagnetic atoms in the sample) and for any small temperatureindependent paramagnetism arising from paramagnetic excited states of the system. N is Avogadro's number, k is the Boltzmann constant, µ is the "magnetic moment" of the molecule, and T is the absolute temperature. By substituting numerical values for N and k, we obtain; X Mcorr 0.125 2 T or 2.83 X Mcorr T 4.2.1 Number Of Unpaired Electrons Once an experimental value of xM has been obtained for a paramagnetic substance, it can be used to determine how many unpaired electrons there are per-molecule or ion. In order to translate the experimental result into the number of unpaired spins, it must be recognized that a measured susceptibility will include contributions from both paramagnetism and diamagnetism in the sample. Even though the latter will be small, it is not always valid to consider it negligible. The most common procedure is to correct a measured susceptibility for the diamagnetic contribution. Compilations of data from susceptibility measurements on a number of diamagnetic materials make it possible to estimate the appropriate correction factors. The diamagnetic susceptibility for a particular substance can be obtained as a sum of contributions from its constituent unit: atoms, ions, bonds, etc. The basic assumption underlying such a procedure, namely, that the diamagnetism associatiated with an individual atom or other unit in independent of environment, has been shown to be valid. The next step is to connect the macroscopic susceptibility to individual molecular moment and finally to the number of unpaired electrons. From classical theory, the corrected or paramagnetic molar susceptibility is related to the permanent paramagnetic moment of a molecule µ, by: xM N 2 2 3RT where N is Avogadro's number, R is the ideal gas constant, T is the absolute temperature, and µ, is expressed in Bhor magnetrons (BM) (1 BM = eh/4 m). Solving this expression for the magnetic moment gives: 3RTX m 2 N 1/ 2 2.84( x M T )1 / 2 As we know, this paramagnetic moment in the spins and orbital motions of the unpaired electrons in the substance. There are three possible modes of coupling between these components spin-spin, orbital-orbital, and spin-orbital. For some complexes, particularly those of the lanthanides, we must consider all three types of coupling. The theoretical paramagnetic moment for such a complex is given by: g[ J ( J 1)]1 / 2 where J is the total angular momentum quantum number and g is the Lande splitting factor for the electron, defined as: g 1 J ( J 1) S ( S 1) L( L 1) 2 J ( J 1) The value of J depends on the total orbital angular momentum quantum number, L, and total spin angular momentum quantum number. 4.2.2 Spin Only Formula For complexes in which spin-orbit coupling is nonexistent or negligible but spin and orbital contributions are both significant, the predicted expression for µ is; [4S (S 1) L( L 1)]1 / 2 This equation describes a condition that is never fully realized in complexes because the actual orbital contribution is always somewhat less than the ideal value. This occurs because the orbital angular momentum is reduced from what it would be in the free metal ion by presence of ligands. In the extreme case, where general situation in complexes having A or E ground states, which would include octahedral d3, d4 (high spin), d6 (low spin), d7 (low spin) and d8 cases. Furthermore, when a complex involves a first-row transition element, even if the ground state is T, the orbital contribution generally may be ignored. For the L=O condition, the above Eq. reduces to; [4S (S 1)1 / 2 2[S (S 1)]1 / 2 which is known as the spin only formula for magnetic moment. By recognizing that S will be related to the number of unpaired electrons (n) by S = n/2, the expression may be further simplified to; [n(n 2)]1 / 2 Check Your Progress - 1 Notes :(i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. 1. Molar susceptibility xM, is given by the relation; xM = .......................................... 2. Magnetic moment, µ is given by the relation; µ = .......................................... 3. The spin only formula is; µ = .......................................... 4. Magnetic moment, µ and number of un-paired electrons, n, are related as; µ = .......................................... 4.3 ANOMALOUS MAGNETIC MOMENTS Table 4.1 indicates that the values of magnetic moment calculated using the spin only formula in number of cases differ from the values obtained from theoretical considerations. This difference is supposed to be due to two reasons, firstly due to the contribution of orbital magnetic moment; and secondly due to dependence of magnetic properties on the experimental temperature (Curie's Law). Table 4.1: Magnetic properties of some complexes of the first-row transition metals Central metal Ti3+ High spin complexes Low spin complexes No. of d No. of No. of µ(expt) µ(calc)b µ(expt) µ(calc)b electrons unpaired unpaired BM BM BM BM electrons electrons 1 1 1.73 1.73 - V4+ 1 1 V3+ 2 2 V2+ 3 3 Cr3+ 3 3 Mn4+ 3 3 Cr2+ 4 4 Mn3+ 4 4 Mn2+ 5 5 Fe3+ 5 5 Fe2+ 6 4 Co3+ 6 4 Co2+ 7 3 Ni3+ 7 3 Ni2+ 8 2 Cu2+ 9 1 1.681.78 2.752.85 3.80390 3.703.90 3.8-4.0 1.73 - - - 2.83 - - - 3.88 - - - 3.88 - - - 3.88 - - - 4.754.90 4.905.00 5.656.10 5.706.0 5.105.70 - 4.90 2 2.83 4.90 2 3.203.30 3.18 5.92 1 1.73 5.92 1 1.802.10 2.0-2.5 4.90 0 - - 4.90 0 - - 4.305.20 - 3.88 1 1.8 1.73 3.88 1 1.8-2.0 1.73 2.803.50 1.702.20 2.83 - - - 1.73 - - - 2.83 1.73 4.3.1 ORBITAL CONTRIBUTION IN MAGNETIC MOMENTS In an octahedral ligand field, only the t2g orbitals remain degenerate and rotationally related. The eg orbitals get separated by 10Dq. Hence, orbital momentum due to the dx2-y2 orbital electron gets quenched, and the spin-only formula should apply. It can be seen that the orbital angular momentum formula should be important for the high spin d1, d2, d6 and d7 ion complexes and low spin d4, d5 ions in octahedral field. In the tetrahedral field, the high spin d 3, d4, d8 and d9 ion should have a significant contribution from the orbital angular momentum. The magnetic moment of [CoCl4]2- (4.4 BM) and that of [Co(H2O)6]2+ (5.0) confirm the above statements, the orbital moment contributes for the high spin octahedral, but not for the tetrahedral complexes. Even for the other ions, where no orbital moment is expected, the observed values significantly depart from the spin-only formula (though the differences are small). This is attributed to the spin-orbitals interactions which oppose the quenching of the orbital moments by mixing the orbitals. This explains the generally, lower µeff values obtained for Cr2+, Cr3+, V3+ and V+ and higher values for spin free Fe2+, Co2+, Ni2+ and Cu2+ complexes. Greatest deviations occur for the Co2+ and Fe2+ complexes, for which unquenched orbital moments contribute significantly. For the 4d and 5d ions, diamagnetism results for even numbered electrons, and paramagnetism to the extent of one unpaired electron only is observed for the old numbered electrons, indicating that spin pairing takes place for these ions as far as possible. This may be due to (i) reduced enterelectronic repulsions in larger sized ions reducing the electron pairing energies, (ii) higher LF or MO splittings. The µ at room temperature is generally lower than µs and cannot be used to determine the unpaired electrons due to (iii) high spin-orbit coupling constants which align L and S vectors in opposite directions destroying the paramagnetism. Further, (iv) the Curie or Curie-Weiss law does not hold, the variation of µ with L is complex and depends upon the number of the electrons present. Some ions like MnO4-, CrO42- and low spin Co3+ complexes show temperature-independent paramagnetism (TIP) even though they do not have any unpaired electron. This is due to the spin-orbit coupling of the ground state to a paramagnetic excited state under the influence of the magnetic field. The degree of mixing is independent of temperature but depends on the applied magnetic field, as the excited state is well separated from the ground state, whose population does not change with temperature. 4.3.2 CURIE'S LAW The observed magnetic moments for the metals in t2g ground state are temperature dependent and usually depart from the µs value due to probably the t2g electron delocalization and lower symmetry ligand field components. Pierre Curie established in 1895 that paramagnetic susceptibility is inversely proportional to the absolute temperature. xM = C/T This expression, which is known as Curie's Law, is actually a restatement of magnetic moment. The Curie law is obeyed fairly well by paramagnetic substances that are magnetically dilute, i.e. those in which the paramagnetic centers are well separated from each other by diamagnetic atoms. In materials that are not magnetically dilute, unpaired spin on neighboring atom may couple with each other, a phenomenon referred to as magnetic exchange. Materials that display exchange behavior can usually be treated with a modification the Curie-Weiss Law; xM C (T ) where ø is a constant with units of temperature. If the interacting magnetic dipoles on neighboring atoms tend to assume a parallel alignment, the substance is said to be ferromagnetic (Fig. 4.1(b)). If, on the other hand, the tendency is for an anti-parallel arrangement of the coupled spins, the substance is anti-ferromagnetic.(Fig. 4.1(c)) In any material that exhibits magnetic exchange, the tendency towards spin alignment will complete with the thermal tendency favoring spin randomness. In all cases, there will be same temperature below which magnetic exchange dominates, this temperature is called the Curie temperature (TC) if the type of exchange displayed is ferromagnetic and the Neel temperature (TN) if it is antiferromagnetic. The change in susceptibility as the temperature is decreased below either TC or TN may be quite dramatic. Paramagnetism Ferromagnetism Antiferromagnetism (a) (b) (c) Fig.4.1 Schematic representations of magnetic dipole arrangements in (a) paramagnetic, (b) ferromagnetic, and (c) anti-ferromagnetic materials. Fe(Phen)2(CNS)2 is an example which shows significant variation in magnetic moment with temperature. (Fig. 4.2) Fig.4.2 The magnetic moment of Fe(phen)2(NCS)2 as a functions of temperature. 4.4 MAGNETIC EXCHANGE COUPLING As we know, a number of transition metal ions form both high and low spin complexes, and we have now seen that magnetic susceptibility allow us to experimentally distinguish one from the other. Within ligand field theory, these two spin configurations in octahedral complexes are explained in terms of relative magnitudes of and pairing energy (P): We associate high spin complexes with the condition P and low spin complexes with P . For complexes in which the energy difference between and P is relatively small, an intermediate field situation, it is possible for the two spin states to coexist in equilibrium with each other. Consider the Fe2+ ion. At the two extremes, it forms high spin paramagnetic [Fe(H2O)]2+ (S=2) and low spin diamagnetic [Fe(CN)6]4- (S=0). Octahedral complexes with 4, 5, 6 or 7 d electrons can be either high-spin or low-spin, depending on the magnitude of the ligand-field splitting, . When the ligand-field splitting has an intermediate value such that the two states of the complex have similar energies, the two states can coexist in measurable amounts at equilibrium. Many "crossover" systems of this type have been studied. 4.5 SPIN CROSSOVER With the change in field strength, change in the magnetic moment i.e., the change from high-spin to low-spin can be explained in terms of splitting of electronic states with the field strength, e.g. the Tanabe-Sugano diagram to these d6 complexes show that near the crossover point between weak and strong field the difference in energy between the spin-free (5T2g) and spin-paired (1A1g) ground states becomes very small (Fig. 4.3) within this region, it is reasonable to expect that both spin state may be present simultaneously and that the degree to which each is represented will depend on the temperature ( - P = kT). A complex illustrating these effects is [Fe(phen)2(NCS)2] (Fig. 4.2). At high temperature a moment consistent with four unpaired electrons is observed, but as the temperature is decreased, a sharp drop in magnitude is observed at 175K where the low-spin form becomes dominant. Usually spin transitions occur somewhat more gradually than in the case shown here, and reasons for the abruptness observed for this complex, as well as some residual paranagnetism seen at low temperature have been discussed extensively. 5 T2g 1 A1g E Fig.4.3 Variation in energies of 5T2g and 1A1g terms with increasing for d6 octahedral complexes. At weak field (high spin complexes) the ground term is 5T2g, while at strong fields (low spin complexes) it is 1A1g Note that in the region immediately on each side of the spin crossover point, the energy difference between the two terms is small; thus high and low spin complexes coexist. In solutions, these systems are fairly straightforward; the change in magnetic susceptibility with temperature can be interpreted in terms of the heat of conversion of one isomer to another. However, treatment of the system as an equilibrium between two spins yields H=3.85 kcal mol-1 and S = 11.4 for the high spin low spin conversion. On the other hand, spin crossover in solids is a complex phenomenon because of cooperative structural changes and changes in the energy separation of the high-spin and low-spin states with temperature. Thus the magnetism of Fe(phen)2(NCS)2 change sharply at 174K, as shown in Fig. 4.2 Check Your Progress - 2 Notes :(i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. (A)(i) Orbital Contribution in magnetic moment is important for high spin............................ions complexes; and low spin ................... ions in octahedral field. (ii) The greatest deviation in magnetic moment occurs for...............complexes. (B) Curie's Law state that..................................................................... ............................................................................i.e. Xm =............ (C) In octahedral complexes for dn configurations (n=...................) the two states (low-spin and high-spin) of complexes can coexist in measurable amount's at equilibrium at ligand field splitting has. (D) The change from high spin to low spin can be explained in terms of................................................................................................... (E) The crossover point is reached when the difference in energy between..........................................................states become very small. 4.6 LET UP SUM UP Substance may be diamagnetic or paramagnetic when any substance is placed in an external magnetic field, there is an induced circulation of electrons producing a net magnetic moment in opposition to the applied field. This is the diamagnetic effect and it arises from paired electrons within a sample. Paramagnetism is produced by unpaired electrons in a sample. The spin and orbital motion of these electrons give rise to permanent molecular moments that tends to align themselves with an applied field. When a substance is subjected to a magnetic field, H, a magnetization I, is induced. The ratio of I/H is called volume susceptibility, k. The volume susceptibility is simply related to the 'gram susceptibility', x and the molar susceptibility, XM asX K d or X M K M d where d and M are the density and molecular weight of the substance, respectively. xM is the molar susceptibility which has been corrected both for the diamagnetic contribution to the susceptibility and for any smll temperature-independent paramagnetism from paramagnetism excited states of the system, and may be given as, X Mcorr 0.125 2 or 2.83 X Mcorr T T when values of Avogadro's number, A, Boltzman constant K, the magnetic moment of the substance, µ and absolute temperature are substituted. Once an experimental value of XM has been obtained for a paramagnetic substance, it can be used to determine how many unpaired electrons there are per molecule or ion. For complexes in which spin-orbit coupling is nonexistent or negligible but spin and orbital contributions are both significant µ is given by; μ [4 S(S 1) L(L 1)]1/ 2 When a complex involves a first row transition element, even if the ground state is T, the orbital contribution generally may be ignored, and we get L=O and µ is given by spin only formula; μ [4 S(S 1)1/ 2 2[S(S 1)]1/ 2 By recognizing that S will be related to the number of unpaired electrons (n) by S = n/2, the above expression is simplified to; μ [n(n 2)]1 / 2 Magnetic moment calculated using the spin only formula in number of cases differ from the values obtained from theoretical considerations. This deviation may due to be either the contribution of orbital magnetic moment, or due to dependence of magnetic properties on the experimental temperature, (Curie's Law). The orbital angular momentum formula may be important for the high spin d1, d2, d6 and d7 ion complexes, and low spin d4, d5 ions in octahedral field. The observed magnetic moments for the metals in t 2g ground state are temperature dependent and usually depart from the µs values due to probably the t2g electron delocalization and lower symmetry ligand field components. The Curie's Law states that paramagnetic susceptibility is inversely proportional to the absolute temperature; xM = C/T If the interacting magnetic dipoles on neighboring atoms tend to assume a parallel alignment, the substance is said to ferromagnetic, and if, on the other hand, the tendency is for an anti-parallel arrangement of the coupled spins, the substance is anti-ferromagnetic. Fe(phen)2(CNS)2 is an example which shows significant variation in magnetic moment with temperature. A number of transition metal ions form both high and low spin complexes. These two spin configurations in octahedral complexes are explained in terms of relative magnitudes of and pairing energy (P). High spin complexes are formed when < P and low spin when > P. For complexes in which the energy difference between and P is relatively small an intermediate field situation, it is possible for the two spin sates to co-exist in equilibrium with each other. Variation in energies of 5T2g and 1A1g terms with increasing for d6 octahedral complexes show, at weak fields (high spin complexes), the ground term is 5T2g, while at strong fields (low spin complexes) on each side of the spin crossover point, the energy difference between the two terms is small; thus high and low complexes may coexist. 4.7 1. CHECK YOUR PROGRESS: THE KEY (i) XM KM d (ii) = 2.84 (XMT)½ (iii) = [4S (S+1) ]½ = 2 (S(S+1)]½ (iv) = [n (n+2)]½ 2.A (i) High spin d1, d2, d6 and d7 ion complexes, and low spin d4 and d5 ions. (ii) For Fe2+ and Co2+ complexes. B. Curie's Law states that paramagnetic susceptibility is inversely proportional to the absolute temperature, i.e. XM= C/T C. For dn configurations (n = 4, 5, 6, 7) the ligand field splitting has an intermediate value. D. In terms of splitting of electronic states with the fields strength. E. Between spin free 5T2g and spin paired 1A1g ground states become very small. 5 METAL π COMPLEXES Structure 5.0 Introduction 5.1 Objectives 5.2 Metal Carbonyls 5.3 5.4 5.2.1 Classification 5.2.2 Isolobal Concept 5.2.3 Methods of Preparation and Properties 5.2.4 Structure 5.2.5 Vibrational Spectra Metal Nitrosyls 5.3.1 Neutral NO and NO- Complexes 5.3.2 Complexes of NO+ 5.3.3 Pure Nitrosyl Complexes 5.3.4 Nitrosyl Carbonyl Complexes 5.3.5 Nitrosyl Halide Complexes 5.3.6 Nitroso Cyanide Complexes Dinitrogen Complexes 5.4.1 5.5 Fixation of Nitrogen Dioxygen Complexes 5.5.1 Heme Proteins and Transportation of O2 5.5.2 Haemoglobin 5.6 Tertiary Phosphine as Ligand 5.7 Let Us Sum Up 5.8 Check Your Progress: The Key 5.0 INTRODUCTION π-bonding in complexes was proposed for the first time, by Pauling (1924), in the form of back-bonding (M L) to account for electro- neutrality of metal to ligand bond. According to him, if the ligand, linked with the metal ion through LM, ϭ-bond, has vacant π-orbitals, it can accept lone pair of electrons from metal-ion (if present) to form ML, πbonds. This also accounts for the extra stability of metal complexes with unsaturated ligands. However the latest and the most successful theory of bonding for metal-complexes Ligand Field Theory (LFT), explained quantitatively while Mπ-bonding stabilizes, the complex, LM π- bonding destabilize it. This also explains positions of CN- and F- ligands in the spectrochemical series. Most transition metals form complexes with a wide variety of unsaturated molecules such as carbon monoxide, nitric oxide, dinitrogen, dioxygen etc. In many of these, the metal is in zero or another low oxidation state and, as we have already mentioned, π-bonding between the metal and the ligands is believed to play an important part in stabilizing these complexes. In this regard metal carbonyls are important as they involve both metal carbon ϭ and π-bonds. In this unit we shall consider the metal carbonyls, anions derived from them, some of their substitution product, and complexes formed by a few other ligands. 5.1 OBJECTIVES The main aim of this unit is to study π-complexes of transition metals, with special reference to bonding and their structures. After going through this unit you should be able to: describe metal carbonyls, their classification, methods of preparation and reactions; with special reference to their structures, discuss how these complexes, almost without exception, conform to the effective atomic number rule and isoloble concept, explain bonding in these complexes in terms of IR spectra; describe preparation, properties and structures of metal nitrosyls, discuss dinitrogen complexes and their importance in the fixation of nitrogen; explain formation of dioxygen complexes with special reference to transportation of oxygen by heme proteins; and describe the nature of complexes with tertiary phosphine as a ligand. 5.2 METAL CARBONYLS The compounds formed by the combination of CO molecules with transition metals are known as metallic carbonyls. Carbon mono-oxide posses a unique property of unsaturation by virtue of which it may combine with a large number of metals under suitable conditions. Such compounds of CO with metals are termed as metallic carbonyls. In carbonyls, a metal atom is directly linked to the carbon atom of a carbonyl group. Since the electrons forming OCM bond are supplied solely by CO molecule, metal atom in carbonyls is said to be in zero oxidation state. In metal carbonyls CO molecules act as neutral ligands. Metal carbonyls vary considerable in their properties ranging from volatile nonpolar to the nonvolatile electrovalent carbonyls. For example-nickel forms volatile nonpolar carbonyls, where as alkali and alkaline earth metals from non-volatile electrovalent carbonyls. The general formula of the carbonyls may be given as Mx(CO)y where M is a metal capable of forming carbonyl. Metal carbonyls may be regarded as parents of number of related compounds such as metal nitrosyl carbonyl, M (NO)y (CO)x, and metal carbonyl hydrides HxM (CO)y. 5.2.1 Classification Carbonyls are classified into two distinct groups: a. Monocular carbonyls: These carbonyls have the general formula Mx(CO)y which contain more than one metal atom per molecule. b. However the carbonyls having 2 metal atoms are called binocular carbonyls, and c. those having more than two metal atoms as ploynuclear carbonyls. Polynuclear carbonyls may be homonuclear e.g. [Fe3(CO)12 or heteronuclear e.g. MnCo(CO)9, MnRe(CO)10] (Table 5.1) They have following characteristics: i. These are almost insoluble in organic solvents. ii. Many polynuclear carbonyls decompose at or below the melting point. 5.2.3 Preparation And Properties Of Carbonyls a. Direct synthesis from metals and carbon mono-oxide, for example: 1. Nickel reacts with CO at room temperature and normal pressure; C Ni 4CO 40 Ni(CO) 4 2. When CO is passed over reduced iron at 108o-220o and pressure of 50 to 200 atom pressure. Fe(CO)5 is formed; Fe - + 5CO Fe(CO)5 Rhenium, osmium and iridium carbonyls could not be prepared by direct reactions. Table 5.1 The binary carbonyls Electrons needed 13 to attain noble gas configuration First transition V(CO)6 series 12 Cr(CO)6 11 10 Mn2(CO)10 9 Fe(CO)5 CO2(CO)8 blue solid white solid yellow solid yellow liquid (sublimes) (m.p.154o) (b.p.103o) Fe2 (m.p.51o) liquid (CO)12 black CO6(CO)16 (b.p.43o) solid black solid Second Mo(CO)6 Te2(CO)10 Ru(CO)5 transition series white white solid colourless Rh2(CO)8* solid(subli liquid mes) 22o) orange solid Ru2(CO)9* Rh6(CO)16 Ru3(CO)12 black solid orange (m.p.- Rh4(CO)12 solid (m.p.-154o) W(CO)6 Re2(CO)10 series white white solid(subli (m.p.177o) Ni(CO)4 orange solid colourless (sublimes) Third transition 8 Os(CO)5 solid colourless liquid Ir2(CO)8 yellow solid Ir4(CO)12 mes) (m.p.15o) yellow solid Os2(CO)9 Ir6(CO)16 red Orange solid solid Os2(CO)12 Yellow solid (m.p.224o) b. Indirect synthesis involving the Gringed reagent: job prepared chromium hexacarbonyl by the action of CO on a mixture of grignard reagent and anhydrous chromium chloride in ether solution. According to Hiber the primary reaction is as follows: C6H5MgBr + CrCl3 + CO Cr(CO)2(C6H5) + MgBrCl + MgBr2 The unstable intermediate compound is composed with acid to yield the hexacarbonyl: 3Cr(CO)2 (C6H5)4 + 6H Cr(CO)6 + 2Cr3+ + 12C6H-5 + 3H2 The reactions gives low yield which can be improved by using high carbon mono-oxide pressure. c. Indirect synthesis involving metal compounds: Metal carbonyls can be prepared by the reaction of CO with certain metal compounds for example: i. Nickel tetracarbonyl may be prepared by passing CO into a suspension of nickel cyanide, sulphide or mercaptide suspended in NaOH solution. 2NiX4 + 2nCO 2Ni(CO)nX + X2 Ni(CO)nX + (4-2n)CO Ni(CO)4 + NiX2 ii.Ruthenium pentacarbonyl may be prepared by the action of CO and Rul3 in the presence of an iodine acceptor: CO ] CO ] RuI 3 [ Ru (CO) 4 I 2 [ Ru (CO) 5 Similarly [Ir(CO)4]7 may be prepared. d. Synthesis by carbonylating the metallic salts with CO in the presence of reducing agent. When salts like R4I3, CrCl3, VCl3 are made to treat with CO in presence of a suitable reducing agent like Mg, Ag, Cu, Na, H2 etc. CrCl3 CO LiAlH y 115 Cr(CO)6 + LiCl + AlCl3 o C 250atmpressure 2Ru(CO)5 + 6Agl 2Rul3 + 10CO + 6Ag 175 210atmpressure C 2Mn2(CO)10+ 2Mgl 2Mnl3 + 10CO + 2Mg 25 e. Synthesis from other carbonyls: when iron pentacarbonyl is exposed to UV light it loses CO and forms Fe2(CO)9. This compound undergoes thermal decomposition to yield iron pentacarbonyl and trimeric tetracarbonyl. .V . Fe2(CO)4 + CO 2Fe(CO)5 U Fe(CO)5 + [Fe(CO)4]3 + CO 2Fe2(CO)9 heat f. Synthesis from Carbonyl hydrides: when iron carbonyl hydride is oxidised by MnO2 or H2O2,[Fe(CO)4]3 is formed. g. By treatment of oxide of metals with CO under pressure: Carbonyls of osmium and rhenium are prepared by the reaction of CO with their oxides under pressure. OsO4 + 9CO C 100 Os(CO)5 + 4CO2 50 atm pressure C Re2(CO)10 + 7CO2 Re2O7 + 17CO 75 atm 200 h. Preparation of Mo (CO)6 and W(CO)6 from Fe(CO)5 MoCl6 + 3Fe(CO)5 Mo(CO)6 + 3FeCl2 + 9CO i. Preparation of Fe2(CO)9 and Os2(CO)9 from Fe(CO)5 and Os(CO)5-9 with cooled solution of Fe(CO)5 and Os(CO)5 in glacial CH3COOH is irradiated with u.v. light, Fe(CO)9 and Os2(CO)9 are obtained respectively. light Fe2(CO)9 + CO 2Fe(CO)5 U.V. .V .light Os2(CO)9 + CO 2Os(CO)5 U Properties of Carbonyls i. The metal carbonyls are crystalline solids, except for nickel carbonyl and the pentacarbonyls of iron, ruthenium and osmium which are liquids. ii. Many are coloured for example: Crystals of cobalt carbonyl are orange and iron pentacarbonyls is yellow oil and nicked carbonyl is colourless. iii. Due to their covalent nature renders them insoluble in water, most of them are soluble in solvents like CCl4. iv. Excepting V(CO)6 all the carbonyls are diamagnetic. V(CO)6 is paramagnetic and its paramagnetic property corresponds to the presence of one unpaired electron. The metal in carbonyls are in zero oxidation state. Table 5.2 Colour And Melting Points Of Some Carbonyls Carbonyl V(CO)6 Melting Point, (oC) Colour and shape Black crystals Decomposes vacuum at 70oC, Sublime in Carbonyl Melting Point, (oC) Colour and shape Cr(CO)6 Colourless crystals Sublime in vacuum Mo(CO)6 Colourless crystals Sublime in vacuum W(CO)6 Colourless crystals Sublime in vacuum Mn2(CO)10 Golden crystals 154o-155o Re2(CO)10 Colourless crystals Sublime at 140o and decompose at 177oC Fe(CO)5 Yellow Liquid B.P. 103oC Fe2(CO)9 Bronze Mica-like Decomposes at 100oC platelets Fe3(CO)12 Dark green crystals Decomposes at 140oC CO2(CO)8 Orange crystals 51oC Ni(CO)4 Colourless Liquid B.P. 43oC Chemical Properties 1. Substitution Reactions: Some or all CO groups present in carbonyls can be replaced by monodentate ligands such as alkyl or aryl isocyanide (CNR) PR3, PCl3, Py, CH3OH etc. Ni(CO)4 + 4CNR Ni(CNR)4 + 4CO Ni(CO)4 + 4PCl3 Ni(PCl3)4 + 4CO Fe(CO)5 + 2CNR Fe(CO)3 (CNR)2 + 2CO 2. Action of NaOH or Na metal: Formation of carbonylate ion: Aqueous alcoholic solution of NaOH reacts with Fe(CO)5 to form carbonylate anion [Fe(CO)4]-. Fe(CO)5 + 3NaOH Na+[H+Fe2-(CO)4]-Na2Co3 + H2O H-atom in [H+Fe2-(CO)4]- ion is acidic which implies that Fe atom in this ion is in -2 oxidation state. Na-metal in liquid NH3 is able to convert Fe2(CO)9. Co2(CO)8, Fe3(CO)12, Cr (CO)6,.Mn2(CO)10 etc, into carbonylate anions and in this conversion these carbonyls are reduced. Fe2(CO)9 + 4Na 2Na 2 [Fe2-(CO)4]2- + CO Co2(CO)8 + 2Na 2Na+[Co-(CO)4]4 3. Action of halogens: Most of the carbonyls react with halogens to yield carbonyl halides. For example: Fe(CO)5 + X2 Fe(CO)4X2 + CO Mo(CO)6 + Cl2 Mo(CO)4Cl2 + 2CO Mn2(CO)10 + X2(X = Br, I) 2Mn(CO)5X Both Co2(CO)6 and Ni(CO)4 are decomposed into metallic halides and CO when treated with halogens. Co2(CO)8 + 2X2 2CoX2 + 8CO Ni(CO)4 + Br2 NiBr2 + 4CO 4. Action of NO: many carbonyls react with nitric oxide (NO) to form metal carbonyls nitrosyls. For example: C Fe(CO)2(NO)2 + 3CO Fe(CO)5 + 2NO 95 3Fe3(CO)9+4NO 2Fe(CO)2(NO)2+ Fe(CO)5+Fe3(CO)12+6CO 5. Action of H2: Formation of carbonyl hydrides (reduction): when Mn2(CO)10 and Co2(CO)8 react with H2, they get reduced to carbonyl hydrides, Mn(CO)5H and Co(CO)4H respectively. C , 200atmpressure 2[Mn-(CO)5H+]0 Mn2(CO)10 + H2 200 C , 200atmpressure 2[Co-(CO)4H+] Co2(CO)8 + H2 165 6. Action of heat: Different carbonyls yields different products when heated for example: C Fe + 5CO Fe(CO)5 250 C 3Fe(CO)5 + Fe3(CO)12 3Fe2(CO)9 70 C 3Fe + 12CO Fe3(CO)12 140 Metal Carbonyls of Different Groups: 1. Carbonyls of Sixth B Metals These form carbonyls of one type only M(CO)6 where M = Cr,Mo, Or W, but chromium also forms Cr(CO)5+ A. Chromium Hexacarbonyl Cr(CO)6. Preparation: i. It is prepared by job's method by passing CO at 50 atm. pressure and at room temperature into a suspension of chromic chloride in ether. Which has been treated with phenyl magnesium bromide at 70oC. ii. Chromium hexacarbonyl can be prepared by treating a solution of a chromic salt dissolved in ether with Al(C2H5)3 and carbon monooxide at a high temperature and pressure. iii. It may also be prepared by carbonylating CrCl3 with CO in the presence of a reducing agent like LiAlH4. C , 200atmpressure Cr.(CO)6 + LiCl + AlCl3 CrCl3 + CO + LiAlH4 175 Properties: 1. Chromium hexacarbonyl exists in colourless rhombic crystals which sublime without decomposition and dissolve in either, chloroform, CCl4 and benzene. 2. It is attacked by air, bromine, cold aqueous alkali, dilute acids conc. HCl and Conc.H2SO4. It is however decomposed by chlorine or by conc. nitric acid. 3. Decompositions: It gets decomposed by F2 at -75oC to form CrF6. 4. Action of Na-Metal or NaBH4: Cr(CO)6 when is treated with Na metal or NaBH4 in liq. NH3 carbonylate anion is formed. In these reactions the carbonyls are reduced. Liq.NH Na 2 [Cr2-(CO)5]2- + CO Cr(CO)6 + 2Na 3 NaBH / Liq . NH Na 2 [Cr2-(CO)10]2- + 2CO Cr(CO)6 4 5. 3 Substitution reactions: Some CO groups present in Cr (CO) 6 can be replaced by pyridine to get a number of products. Py Py py Cr (CO) 3 ( Py) 3 Cr(CO)6 Cr(CO)4(Py)2 Cr2(CO)7(Py)5 Yellow brown solid Yellow red solid Bright red solid B. Molybdenum Hexacarbonyl and Tungsten Hexacarbonyl. Preparation: 1. Both these carbonyls may be prepared by job's method which involve the reaction of either MoCl6 or WCl6 with CO in the presence of phenyl magnesium bromide. 2. Both may also be prepared by the action of CO at 225o and 200 atm. pressure on metallic molybdenum or tungsten reduced in the presence of copper or iron. Properties: 1. They are colourless, Mo(CO)6 sublimes at 40oC and boils at 156.4o, whereas W(CO)6 sublimes at 50oC and boils at 175oC. 2. They are stable in air and dissolve in organic solvents like ether, chloroform, CCl4 and benzene. 3. Mo(CO)6 do not react with air, cold aqueous alkali, acids, except conc. nitric acid or with thiols or nitric oxide. 4. Bromine and chlorine can decompose Mo(CO)6 and W(CO)6. 5. With pyridine, phenanthroline and ethylene diamine, the CO group in Mo(CO)6 and W(CO)6 is replaced. M(CO)5Pyr2 M2(CO)7PYr5 M(CO)3PYr3 M(CO)6 Pyridine 2. Carbonyls or VII Group: These form volatile carbonyls of the formula M2(CO)12 where M = Mn, Te and Re. Manganese carbonyl, Mn2(CO)10. Preparation: 1. This is prepared by treating manganese iodide and magnesium with CO in ether under high pressure. In this reaction, magnesium acts as a reducing agent. C , 210atmpressure Mn2 (CO)10 2MnCl2+10CO+2Mg (in diethyl ehter) 250 + 2MgI2 2. By carbonylating MnCl2 with CO in presence of (C8H5)2CONa C140atm. Mn2(CO)10+ 2MnCl2+10CO+4(C8H5)2CONa 165 4(C6H5)2CO+4NaCl Properties: 1. Manganese carbonyl forms volatile, golden yellow, crystalline, solid which melts at 155oC in a sealed tube. It is soluble in organic solvents. It is slowly oxidised in air, especially in solution. 2. Action of halogens: Mn2(CO)10 reacts with halogens to form carbonyl halides. Mn2(CO)10 + X2 (X=Br2I) 2Mn(CO)5X 3. Action of Na-Metal: Na-metal in liquid NH3 converts Mn2(CO)10 into carbonylate anion. In this reaction the oxidation state of Mn decreases from zero to -1; Liq.NH 2Na [Mn-(CO)5]Mn2(CO)6 + 2Na 3 4. Action of H2: Mn2(CO)10 gives carbonyl hydride, Mn(CO)5H; in the formation of this compound the oxidation state of Mn decreases from zero to -1. 200C , 200atmpressure 2[Mn-(CO)5H+]0 Mn2(CO)10 + H2 5. Substitution Reaction: Mn2(CO)10 reacts with PR3 to form Mn(CO)4(PR3): Mn2(CO)10 + PR3 2Mn(CO)4(PR3) + 2CO 6. Diamagnetic nature: Mn2(CO)10 is a diamagnetic substance, diamagnetic character is confirmed by the fact that all the electrons in Mn2(CO)10 are paired and Mn-Mn bonds is present in it. 3. Carbonyls of VIII Group Metals A. Carbonyls of Iron: Three carbonyls of iron are known, these are: a. Iron Pentacarbonyl, Fe (CO)5 Preparation: i. It can be prepared by the action of CO on iron powder at 200 oC and 200 atm. pressures. Fe + 5CO Fe (CO)5 ii. Recently it has been prepared by the action of CO on Ferrous iodide in the pressure of Cu which acts as a halogen acceptor. 200C Fe (CO)5I2 FeI2 + 4CO iii. It may also be prepared by the action of CO on FeS at 200 oC and 200 atm. pressure in the presence of copper. 200C , 200atmpressure 2Fe(CO)5 + Cu2S 2FeS + 10CO + 2Cu Properties: i. Fe(CO)5 is a yellow liquid which is soluble in methyl alcohol, ether, acetone and C6H6. It is insoluble in H2O. ii. Decomposition: M thermal decomposition at 250oC it yields pure Fe. 250C Fe + 5 Co 2Fe(CO)5 iii. Action of u.v. light: When cooled solution of Fe(CO)5 in glacial CH3 COOH is irradiated with u.v. light, Fe(CO)9 is formed. The above reaction is reversed in darkness. iv. Hydrolysis: Fe(CO)5 gets hydrolysed by H2O and acids Fe (CO)5 + H2SO4 FeSO4 + 5CO + H2 v. Action of alkali: Fe(CO)5 + 4NaOH Na+[Fe2-(CO)4H+]- + Na2CO3 + H2O vi. Action of NH3: with NH3 it yields Fe(CO)4H2 Fe(CO)5 + H2O + NH3 Fe (CO)4H2 + NH2COOH vii. Reaction with halogen: Fe(CO)5 + X2 Fe (CO)4X2 + CO The velocities of these reactions have been found to follow the order CI < Br < I. b. Iron Enneacarbonyl, Fe2(CO)9 When iron pentacarbonyl is dissolved in glacial acetic acid and is exposed to u.v. light for 6 hours, Fe2(CO)9 is formed which dissolves in acetic acid, on cooling with water, golden crystals of the enneacarbonyl are precipitated and are filtered off. 2Fe(CO)5 Fe2(CO)9 + CO Properties i. Fe2(CO)9 forms golden triclinic crystal, it is diamagnetic and nonvolatile. It is insoluble in water but soluble in toluene and pyridine. When heated to 50oC decomposes to form Fe2(CO)12. 3Fe2(CO)9 3Fe (CO)5 + Fe3(CO)12 ii. Action of heat: when heating is done at 100oC, Fe2(CO)9 decomposes to form iron, CO and some Fe (CO)12. 4Fe2(CO)9 Fe + Fe(CO)5 + Fe3(CO)12 + CO iii. Action of NO: With NO it gives Fe(CO)2 (NO)2 together with Fe(CO)5 and Fe2(CO)12. 3Fe2(CO)9+4NO2Fe(CO)2(NO)2+Fe(CO)5 + Fe3(CO)12 + 6CO c. Iron Dodecarbonyl, Fe3(CO)12. Preparation It can be prepared by heating Fe2(CO)9 dissolved in toluene at 70oC. 3Fe2(CO)9 3Fe(CO)5 + Fe3(CO)12 Properties i. Fe3(CO)12 forms deep crystals which are soluble in organic solvents like toluene, alcohol, ether and pyridine. ii. Action of Heat: When heated to 140oC Fe3(CO)12 decomposes to give metallic iron and CO. C 3Fe + 12CO Fe3(CO)12 140 iii. Reaction with Na: Carbylate axion is formed when Fe3(CO)12 reacts with Na metal in Liq. NH3. Liq.NH 3Na2+[Fe2-(CO)4]2 Fe3(CO)12 + 6Na 3 iv. Substitution Reactions: This reaction takes place with pyridine and methyl alcohol. Fe3(CO)12 + 3Py Fe3(CO)9(Py)3 + 3Fe(CO)5 B. Carbonyls of Cobalt It forms two carbonyls i. Cobalt Octacarbonyl, CO2(CO)8 Preparation i. It is prepared by the action of CO and the reduced metallic cobalt at 220oC and 250 atm. 2Co + 8CO Co2(Co)8 ii. When a solution of cobalt carbonyl hydride is treated by an acid, hydrogen is evolved and Co2(CO)8 remains 2Co(CO)4H Co2(CO)8 + H2 Properties 1. Cobalt octacarbonyl forms orange transparent crystals. 2. It is insoluble in water but is soluble to some extent in alcohol, ether, CS, etc. 3. Action of air: On exposure to air, dicobalt octacarbonyl is converted into deep violet basic carbonate of cobalt. 4. Action of Na-metal in liq. NH3: When CO2(CO)8 reacts with Nametal in liq. NH3, it gets reduced to carbonylate anion. NH 2Na[Co(CO)4] Co2(CO)8 + 2Na lig 3 5. Action of NO: Co2(CO)8 reacts with NO at 40oC to form cobalit carbonyl nitrosyl, [Co-(CO)3(NO)]0. Thus in this reaction the oxidation state of cobalt decreases from 0 to -1. Co2(CO)8 + 2X2 2Co2X2 + 8CO 6. Dispropotination Reaction a. Strong bases cause disproportination into Co(+2) and Co(-1) 2Co(CO)8 + 12NH3 2[Co(NH3)6][Co(CO)4]2 + 8CO b. With isocyanides it gives penta-co-ordinate cobalt(I) cation. Co2(CO)8 + 5CNR [Co(CNR)5][Co(CO)4]+4CO (ii) Dodecarbonyltetra Cobalt, [CO4(CO)12] Preparation 1. It is prepared by heating Co2(CO)8 at 60oC. 2. It may also be obtained by oxidizing cobalt carbonyl hydride below -26oC. Properties: i. It is black crystalline solid. ii. It is very unstable easily oxidized by air and can be recrystallized from hot benzene. C. Carbonyls Of Nickel. (i) Nickel Tetracarbonyl, Ni(CO)4 Preparation i. Ni(CO)4 can be prepared by the action of CO on reduced nickel at 30-50oC. Ni + 4CO Ni(CO)4 ii. When Nickel iodide is heated with CO in the presence of a halogen acceptor, nickel carbonyl is formed. NiI2 + 4CO Ni(CO)4 + I2 Properties i. It is colourless liquid, m.p. = -23oC, b.p. = 43oC ii. It has no solubility in water but dissolved in organic solvents. iii. It decomposes at 180o-200oC in to nickel and CO. C Ni + 4CO Ni(CO)4 180 3 iv. It reacts with H2SO4 and form NiSO4 Ni(CO)4 + H2SO4 NiSO4 + H2 + 4CO v. It reacts with Ba(OH)2 and gives BaCO3 Ni(CO)4 + Ba(OH)2 H2Ni(CO)3 + BaCO3 D. Carbonyls of Ruthenium It forms three carbonyls : a. Ruthenium Pentacarbonyl, Ru(CO)5 Preparation i. It is prepared by the action of CO and reduced ruthenium at 200oC and 200 atm. pressures Ru + 5CO Ru(CO)5 Properties i. It is colourless soluble liquid having m.p. = -22oC ii. It has no solubility in water but is soluble in alcohol, benzene and CHCl3. iii. It undergoes decomposition to give Ru2(CO)9 and Ru3(CO)12. iv. It reacts with halogen to yield Ru(CO)Br and CO. v. It is photosensitive and yields ruthenium enneacarbonyl. b. Ruthenium Enneacarbonyl, Ru2(CO)9 It is prepared by exposing pentacarbonyl to u.v. radiation. It forms yellow monoclinic crystals. It is volatile; it is less stable towards heat, with iodine. It yield, Ru (CO)2I2. c. Ru3(CO)12 It is prepared in small quantities along with Ru 2(CO)9 when Ru(CO)5 is heated at 50oC or by exposing Ru(CO)5 to u.v. light. It is a green crystalline solid. E. Carbonyl Of Osmium It forms two carbonyls: a. Osmium pentacarbonyl. Os2(CO)5 It is a colourless having m.p.-15oC. It is obtained. i. by the action of CO on OsI3 at 120oC and 200 atm. pressure in the presence of copper. ii. by the action of CO on OsO4 at 100oC and 50 atm. pressure. OsO4 + 9CO Os(CO)5 + 4CO2 b. Osmium eneacarbonyl, Os2(CO)9 It is a yellow crystalline solid. It is prepared by the reaction of OsI3 with CO in the presence of copper, it is more stable towards heat than Ru4(CO)9. It melts at 224oC and sublimes without decomposition. F. Carbonyl Of Iridium It forms 2 carbonyls: a. Iridium Octacarbonyl, Ir2(CO)8 It is prepared by the reaction of either KIr2Br6 or KIr2Br6 or KIr2I6 with CO at 200oC and 200 atm. pressures. It is yellow crystalline solid having m.p. 160oC. b. Iridium Dodecarbonyl, Ir4(CO)12 It forms orange yellow rhombohedra crystals which decomposes at 200oC. It is prepared by treating Irl3 with CO under pressure. G. Carbonyl of Platinum Preparation i. When CO is passed over PtCl2 at 250oC, PtCl2(CO) and 2PtCl23CO are obtained on heating these yield PtCl2(CO)2. 3PtCl2 + 5CO PtCl2.2CO + 2PtCl2.3C0 Properties These carbonyls are decomposed by water and HCl. PtCl2.CO + H2O Pt + 2HCl + CO2 PtCl2.CO + H2O Pt + 2HCl + CO2 + CO PtCl2.CO + HCl H[PtCl3.CO] PtCl2.CO + HCl H[PtCl3.CO] + CO 5.2.4 Structure of Metal carbonyls 1. Effective Atomic Number Rule: The structure of CO is :C:O: It is probable that the lone pair of electrons on the carbon atom can be used by forming a dative bond with certain metals (MC O, Thus (MC O) types of bonds were assumed to be present in metal carbonyls. In the formation of MC O bonds, the electrons are supplied by the molecules of CO and the metal atom is thus said to have zero-valency. The Number of molecules of carbon mono-oxide which can unite with one atom of the metal is controlled by the tendency of the metal atom to acquire the E.A.N. of the next inert gas. For the stable nonnumeric carbonyl. E.A.N. = m + 2y = G Where M = Atomic number of the metal M Y = No. of CO molecules G = At. No. of next inert gas Carbonyls Cr(CO)6 Atomic Number of the metal 24 Number of electron E.A.N. Succeeding contributed by CO inert gas groups 12 36 Kr(36) Fe(CO)5 26 10 36 Kr(36) Ni(CO)4 28 8 36 Kr(36) Mo(CO)6 42 12 54 Xe(54) Ru(CO)5 44 10 54 Xe(54) W(CO)6 74 12 86 Rn(86) Os(CO)5 76 10 86 Rn(86) on the basis of E.A.N. rule it can be explained why Ni atom fails to form a hexacarbonyl Ni(CO))6 because EAN or Ni atom in Ni(Co)6 would be equal to 28 + 2 x 6 = 40. Which is not the atomic number of any of the noble gases. Mononuclear carbonyls having the metallic atom with odd At. No. V(CO)6 and Mn(CO)5 & Co(CO)4 are the example of such carbonyls. They do not obey EAN rule V = 23e6CO = 12 eV(CO)6= 35e- Mn = 25 e5CO = 10 eMn(CO)5= 35e- Co = 27 e4CO= 8 eCo(CO)4= 35e- Therefore the metals with odd atomic number cannot form monocular carbonyls but forms polynuclear carbonyls for example, Mn(25) and Co(27) form polynuclear carbonyls. 2. Polynuclear Carbonyls: Sidgwick and Bailey gave the general formula for polynuclear carbonyls. G Where X m 2y X 1 X G = The At. No. of next Inert Gas. M = The At. No. of metal atom. Y = The No. of CO molecules in one molecule of the carbonyl. Mn2(CO)10, CO2(CO)8 etc. obey the E.A.N. rule, their E.A.N. per atom of metal is 36. For example: E.A.N. of Mn2(CO)10 may be calculated as: Electrons from 2Mn Atom = 25 x 2 = 50 Electron from 10CO molecules = 10 x 2 = 20 Electrons from one Mn-Mn Bond= 1x2=2 Total = E.A.N. for one Mn atom = 72/2 = 36 72 The formation of binuclear carbonyls having metal atoms with odd atomic number can also be explained on the basis of 18-electron rule as shown below for Co2(CO)8. Co2(CO)8 2Co = 2 x 9e- = 8Co = 2 x 8e- 18e= 16e- Co-Co bond = 1x 2e- = 2eCo2(CO)8 = 36e Electrons on one Co atom = 18eDrawback X-Ray diffraction method shows that the bonds are intermediate between the M-C = 0 and M=C=0 states, i.e. there is some double bond character in M-CO. The EAN rule does not explain double bond character. This is explained by both MOT and VBT. 3. Molecular Orbital Approach According to the M.O.T. carbon and Oxygen atom undergo overlapping to form bonds in CO as follows i. 2 sp hybrid orbital of carbon and 2px of oxygen overlap to form a localised bond. ii. 2py of carbon and 2py of oxygen overlap to form a π-bond. iii. 2pz of carbon and 2 pz of Oxygen overlap to form another πbond. iv. There will be 2 non-bonding electrons in the 2sp hybrid orbital of carbon. v. There will be 2 non-bonding electrons in 2s atomic orbital of oxygen. vi. There will be no electron in the anit-bonding molecular orbitals, formed as result of anti π overlapping. As the total No. of bonding electrons is six and that of antibonding electrons nil, bond order of the molecule is three. Hence, the No. of bonds between carbon and oxygen atoms in CO molecules is 3, one and two π. The lone pair of electrons on carbon could be expected to form a strong dative bond ( ) due to the electron density remaining close to the nucleus of the carbon atom. As metal atom-carbon mono-oxide bonds are readily formed in metal carbonyls. It is expected that there is some additional bonding mechanism in the formation of metalcarbon monooxide bonds in the metal carbonyls. Mechanism 1. Firstly, there is a dative overlapping of filled carbon -orbital i.e. 2sp hybrid orbital with an empty metal -orbital (MCO) as in the figure 5.1. - m + + C = 0 : - M + C = O: Fig. 5.1 L - M bonding 2. Secondly, there is a dative overlapping of a filled d-orbitals of metal with empty antibonding p-orbital of the carbon atom (MCO), resulting in the formation of a dative π bond. The shaded portion in figure, indicate the filled orbitals, whereas empty portions indicate vacant orbitals. i.e. having no electrons. As there is a drift of metal electrons into CO (MCO) orbitals will tend to make the CO as a whole negative and at the same time there is a drift of electrons from CO to the metal (MCO) to make CO positive. Thus enhancing the acceptor strength of the π bond formation and vice versa. Fig. 5.2(a) dπ - Pπ back bonding Fig. 5.2(b) M - CO and π bonding 3. Valence Bond Method: Monocular Carbonyls In this method, the molecule may be represented by resonance structures. M -+ C – O M = C = O with a large amount of the double bond character, it is this structure that account for their stability. From either the molecular orbital or the valency bond view point, back donation is seen in both. Structure of Ni(C)4 1. The vapour density of nickel carbonyl and the freezing points of its solution in benzene indicate the molecular formula to be Ni(CO)4. 2. Electron diffraction studies shows that Ni (CO)4 molecule has tetrahedral shape with Ni-C-O linear units. Figure shows that the Ni-C bond length in this molecule is 1.50Ao which is shorter by 0.32Ao in comparison in Ni-C single bond length (=1.82Ao) found in carbonyls. The C-O bond length in this carbonyl has been found to equal to 1.15 Ao. Which is larger that the C-O bond length in CO molecule (=1.128Ao) (Fig. 5.3) 3. Fig. 5.3 L Tetrahedral structure of Ni (CO)4 molecule Raman Spectra shows that nickel atom in the nicle carbonyl must be tetrahedrally hybridised as in the figure 5.3. Titrahedral shape of Ni (CO)4 arises due to Sp3 hybridisation of Ni- atom. Which is diamagnetic, all the ten electrons present in the valence shell of Ni atom are paired in 3d orbitals. Thus the valence shell configuration of Ni atom in Ni (CO)4 molecule becomes 3d10 4SO CO Ni bond is caused by the overlap between the empty sp3 hybrid orbital on Ni-atom and doubly filled sp hybrid orbital on C atom in CO molecule, as in the figure 5.3 (b) Because of the formation of 4 OC M bonds, a large negative charge gets accumulated on central Ni atom. Pauling suggested that the double bonding occurs with the back donation of d-electron from Ni atom to CO ligands to such an extent that electroneutrality principle is obeyed. According to which the electron pair is not shared equally between Ni and C-atoms of CO ligand but gets attracted more strongly by C-atom which prevents the accumulation of negative charge on Ni-atom, in keeping with the greater electronegativity of C-atom compared to Ni atom. Evidences: 1. The above structure (Fig. 5.3) is supported by the following reactions: i. When an alcoholic solution of the carbonyl is treated with orthophenanthroline to yield a stable ruby-red compound Ni(CO)2 phen. It confirms that two C=O groups of Ni(CO)4 are replaced by one molecule of phenanthroline. ii. Similarly the reactions of Ni(CO)4 with diarsine indicates the two C=O groups are replaced and remaining two are retained. 2. i. Structure of Fe(CO)5: The various evidences are: The vapour density and the freezing points of benzene solution shows that its molecular formula is Fe(CO)5. ii. Electron diffraction, Raman and I.R. spectra shows that it has trigonal bipyramidal shape and Fe-C axial bond and Fe-C basal bond lengths are equal to 1.797Ao and 1.842Ao respectively. It has dsp3 hybridisation of Fe atom (Fig. 5.4(c). III. Molecule is Diamagnetic and the distance Fe-C is 1.84Ao(Fig. 5.4). Fig. 5.4 (a) : Structure of Fe(CO)5 Structure of Cr(CO)6 It has octahedral configuration. The internuclear bond lengths are: Cr-C Cr-O C-O 1.92 3.08 1.16Ao According to old concept when chromium forms Cr(CO)6 one electron of 4s orbital missing and three 4p orbitals become empty which are hybridised to form six d2sp3-hybrid orbitals six molecules of CO donate a lone pair of electrons each to six vacant hybrid orbitals to form six CrCO -bonds as shown in the Figure 5.5. Therefore Cr(CO)6 molecule is diamagnetic in nature and octahedral in geometry. When chromium atoms form chromium carbonyl [Cr(CO)6] the metal atom exhibits d2sp3 hybridisation. Out of 6 d2sp3 hybrids three hybrid orbitals are half filled and three hybrid orbitals are empty. Three electrons remain in 3d orbitals as shown in the figure. 5.5(a) and (b). The Bond Structure of Cr(CO)6 Shows 2 kinds of bonds between Cr and Co. (a) Simple Covalent Bonds Cr-C 0 (b) Double bonds Cr c=O In the resultant resonance structure all Cr-C bonds have been identical, each of the 6 CO groups get linked to the metal atom by a bond are constructed from the d-orbitals of the metal atom. CO (groups I) are bound to the metal atoms by simple ionic bonds. Fig. 5.5 Structure of Cr(CO)6 Hence these CO groups are replaceable by any other molecule capable of donating lone pair of electrons to the metal atom where as CO (groups II) are not replaceable. In the same way structure of MO(CO)6 and W(CO)6 can be explained. Various internuclear bond lengths of these carbonyls are as under: TABLE : INTERNUCLEAR BOND LENGTHS Metal M-C(A) M-O(A) C-O(A) Cr 1.916 3.98 1.171 Mo 2.063 3.23 1.145 W 2.06 3.19 1.148 Thus, the bond structure of Cr(CO)6 shows 2 kinds of bonds between Cr and CO. i. Simple covalent bonds ii. Double bond Cr-C 0 (I) Cr = 0 (II) Structure of Polynuclear Carbonyls These crabonyls obeys EAN rule, if two electrons from each metal metal bond present in these carbonysls are included in calculating the electrons per metal atom; eg metal-metal bonding is evident in Mn2(CO)10 as in Figure. 5.6 Structure of Dinuclear Carbonyls (a) (i) Mn2(CO)10 : Its structrue is shown in Fig 5.6. Fig. 5.6 (ii) Structure of Fe2(CO)8 I.R. and X-Ray study show that in this molecule each Fe atom is directly linked with the other Fe atom by a S-bond (Fe-Fe S-bond) to three bridging carbonyl gropus (>C = 0) by a bond (Fe-C bond) and to three terminal carbonyl gropus (-C = 0) by a co-ordinate bond (FeC co-ordinate bond). The presence of Fe-Fe bond is supported by the diamagnetic character of Fe2(CO)9 molecule. Fe-Fe bond distance has been found to be equal to 2.46Ao. The terminal C-O bond distances from the structure given in figure. (5.7) The co-ordination number of each Fe atom is not equal to 6 but equal to 7. Fig. 5.7: Structure of Fe2(CO)8 Similarly structures of Co2(CO)8 can be represented as in Fig. 5.8 Fig. 5.8: Structure of Fe2(CO)8 (b) Structure of Trinuclear Carbonyls Os3(CO)12 and Ru3(CO)12 possess similar structure (Fig. 5.9a) where as Fe3(CO)12 has a different structure 5.9(b). Os and Ru molecules do not have any bridging CO group (Fig. 5.9 (a)). In Fe3(CO)12 each of the two Fe atoms is linked with three terminal CO groups, two bridging CO groups and third Fe atom is linked with four terminal CO groups and to each of two Fe atoms.(Fig. 5.9 (b). It is also shown by a structure similar to Fig. 5.9. Structure of Fe3(CO)12 According to old concept each iron atoms gets hybridized trigonal bipyramidally (dsp3). The three trigonal bipyramides get arranged in such a manner so that the carbonyl groups at two of the equatorial apices of each bipyramid and held in common by two bipyramides dxz and dyz orbitals are available to form Fe-Fe bonds. It is solid. The three Fe atom get situated at the corner of an isosceles triangle and the twelve CO arranged at the twelve CO arranged at the vertices of an icosahedra. Two Fe-Fe bond lengths are 2.698 Ao and one Fe-Fe bond length is 2.56Ao. (Fig. 5.9) Fig. 5.9 Fe3(CO)12 Complex Fig. 5.9 (a) M3 (CO)12 Structure Fig. 5.9 (b) Structure of Fe3 (CO)12 (c) Tetra and Hexanuclear Carbonyls On the similar grounds structures of tetranuclear carbonyls, such as M4(CO)12 [M = Co, Rh, Ir] and hexanuclear carbonyls, M6(CO)16 eg. Rh6(CO)16 can be represented as in Figs. 5.10 and 5.11 respectively. Some heteronuclear carbonyls are also known e.g. Mn2Fe(CO)14 is shown in Fig. 5.12. (a) (b) Fig. 5.10 (a) Structure of Ir4(CO)12 (b) M4(CO)12; M = Co or Rh Fig. 5.11 Structure of Rh6(CO)16 Fig. 5.12 Structure of Mn2Fe(CO)14 5.2.5 Vibrational Spectra IR spectra give important information regarding nature of carbonyl groups present in metal carbonyl complexes. We can differentiate between the terminal carbonyl e.g. in Mn2(CO)10 and bridging carbonyl groups, as in Co2(CO)8. Metal-carbon distances in Fe2(CO)9 and Co2(CO)8 fall into two groups, metal-bridging carbonyl distances being about 0-1 A longer than metal-terminal carbonyl distances. Such a difference is compatible with the concept of two-electron donation by terminal carbonyls, and one-electron donation (to each of two metal atoms) by bridging carbonyls, through the possible existence of bonds of different strengths makes quantitative interpretation impossible. That the extent of bonding to terminal and bridging carbonyls is different is clearly shown by carbonyl stretching frequencies. Carbon monoxide itself has stretching frequency of 2143 cm-1; neutral metal carbonyls known to have no bridging carbonyl groups have stretching frequencies in the range 2125-2000 cm-1; and Fe2(CO)9 and Co2(CO)8, in addition to showing bands in this region, also show carbonyl absorption at 1830 and 1860 cm-1 respectively. In general, carbonyl absorption in the 1900-1800 cm-1 region is indicative of the presence of bridging carbonyl groups in uncharged species, though the presence of other groups may result in the lowering of the stretching frequencies of terminal carbonyl groups into this region (in carbonylate anions such as [Co(CO)4]and [Fe(CO)4]2- very low carbonyl stretching frequencies of 1883 and 1788 cm-1 respectively result from the strong metal-carbon bonding which stabilizes the low oxidation state of the metal. In a few neutral species believed to contain carbonyl groups bonded to three metal atoms, stretching frequencies of 1800 cm-1 or less are found. Thus, in summary the terminal carbonyl absorption is obtained in the range of 2125-2000 cm-1, while bridging carbonyl frequency is obtained in the 1900-1800 cm-1 region. While, the strong metal carbon bonding is indicated by very low carbonyl structing frequencies of 1883 and 1788 cm-1 respectively. Check Your Progress-1 Notes :(i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. (a) Generally metal carbonls involve............................... -and ......... ..................... -bonding between CO and metal atom. (b) Metal atoms with even number of electrons easily form............carbonyl, but the metal ions with odd number of electrons give.......................or............................... (c) I.R. spectra of carbonyls at..........................cm-1 for show the C-O terminal stretching carbonyl frequency group and at................cm-1 for the bridging carbonyl group. The M-CO π bond is indicated by the absorption at...................and.............Cm-1 respectively. (d) While Mn2(CO)10 has........................bridging carbonyl group, Fe2(CO)9 has................................................................................ 5.3 METAL NITROSYLS Nitrosyls are the compounds in which the nitrogen of the nitrosyl group is directly bonded to the atoms or ions, or the compounds containing nitric oxide group are called nitrosyl compound. NO molecule is an odd electron molecule having an unpaired electron, it readily unites with other elements by direct addition to form nitrosyl compounds. Nitric oxide form nitrosyl compound by the following 3 ways: A positive ion, NO+ is formed due to the loss of an electron which i. then combine with atom or molecule (:N:::O)+ or (:N O:)+ A negative ion NO- is formed due to the gain of an electron from ii. some electropositive metal and it has structure as below: (:N:: O)- or (:N=O:)iii. NO may act as a co-ordinating group through the donation of an electron pair, such behaviour involve neutral molecule or NO + or NO- group. The electronic configuration of NO group is so flexible that it is rather impossible to write its any one configuration in metal nitrosyls. However in all nitrosyls nitrogen atom is linked with the metal possible modes are given below: I :N :N :O: Links :O: Links II III with bonds (neutral) bonds D :N V D :N :O: with Links and IV :N :O: with Links :O:with Accepts dative bonds dative bonds electron (cationic) (cationic) from metal (cationic) (anionic) Mode (I) is rarely seen, while (II), (III) and (IV) modes are formed after transferring one electron to the metal atom. Out of these modes (II) and (III) are similar to the carbonyl group linked with a metal atom. Mode (V) is seen only in a few complexes only, e.g. [Co(CN)5NO]. 5.3.1 Neutral NO and NO- Complexes As has been pointed out metal complexes of neutral NO and its anion, NO- are very rare. Fe(NO)2(CO)2 is supposed to be the important example of metal complex with neutral nitric oxide molecule. This is prepared by the action of nitric oxide on Fe(CO)5. During the reaction, NO replaces neutral CO, hence it is supposed to be a complex of neutral NO. However, the experimental evidences are not supportive. The important examples of anionic NO- are the metal complexes, formed by the action of nitric oxide with ammonical solution of Cobalt (II) salts, with the general formula [Co)NH3)5NO]X2. Two series of isomeric complexes are formed one having black colour, while the other one has red colour. The black series contains the monomeric cation [Co(NH3)3NO]2+, in which a very low N-O stretching frequency of 1170 cm-1 and a long N-O bond (variously reported as 1.26 or 1.41A) suggests the presence of NO-. The red series are derivatives of hyponitrite, the structure of the diametric cation being. Similarly [Co(CN)5NO]3- anion is also supposed to be a complex of NO- anion, since it gives NO- stretching frequency at 1150 cm-1. 5.3.2 Complexes of NO+ Most complexes of nitric oxide and transition metals are best considered to be those of the NO+ ion, three electrons being transferred to the metal atom: M-N back π-bonding then takes place in exactly the same way as for carbon monoxide. Because of its positive charge, however, coordinated NO is a better π-acceptor than coordinated CO, and the N-O stretching frequency in complexes of NO+ is some 300-500 cm-1 lower than that in salts such as NO+BF4-. Two NO+ derivatives of iron may be mentioned briefly here. The species formed in the brown-ring test for nitrate is [Fe(H2O)5NO]2+. The equilibrium [Fe(H2O)6]2+ + NO [Fe(H2O)5NO]2+ + H2O is reversible, and the brown complex may be destroyed by blowing nitrogen through the solution to remove nitric oxide. In this species the N-O stretching frequency is 1745 cm-1, and the magnetic moment is 3.9 B.M., corresponding to the presence of three unpaired electrons; formally, therefore, the ion is a high-spin d7 complex of Fe1 and NO+, but the N-O stretching frequency indicates very strong π-bonding and the intense brown colour strongly suggests Fe1-NO+ charge transfer. 5.3.3 Pure Nitrosyl Complexes Pure nitrosyl complexes of M(NO)4 formula have been reported. Important complexes in this series are Fe(NO)4, Ru(NO)4 and Co(NO)4. In addition to this trinitrosyl cobalt, Co(NO)3 has also been reported. Fe(NO)4 is prepared by the action of nitric oxide under pressure and below 45oC temperature on Fe(CO)5. While M(NO)4 nitrosyls of Ru and Co are prepared by the same method using Ru2(CO)9 and Co2(CO)8 respectively. Fe(NO)4 is a black crystalline substance which decomposes in to Fe(NO) and Fe(NO)2. The structure of tetranirtrosyl iron, Fe(NO)4 has been shown tetrahedral, while that of trinitrosyl cobalt, Co(NO) 3 pyramidal. Nitric oxide links with iron, following II mode, as a three electron donor and results in a strong ML back π-bonding (Fig. 5.13). 5.3.4 Nitrosyl Carbonyl Complexes Mononuclear nitrosyl carbonyls are restricted to the following compounds; Co(NO)(CO)3, Fe(NO)2(CO)2, Mn(NO)3CO and Co(NO)3 (isoelectronic with Ni(CO)4; Mn(NO)(CO)4 (isoelectronic with Fe(CO)3; and V(NO)(CO)5 (isoelectronic with Cr(CO)6). In addition a binuclear species Mn2(NO)2(CO)7 (isoelectronic with Fe2(CO)9 and a number of nitrosyl complexes containing organic groups or triphenylphophine as substituents have been prepared. Nitric oxide displaces carbon monoxide from V(CO) 6, (Ph3P)2Mn2(CO)8, Fe2(CO)9 and Co2(CO)8 to give V(NO)(CO)5, Mn(NO)(CO)4, Fe(NO)2(CO)2 and Co(No)(CO)3 respectively; the further action of nitric oxide on the manganese and cobalt compounds yields Mn(NO)3(CO) and Co(NO)3. All of these substances are solids of low melting point or liquids which are thermally rather unstable and are decomposed by air and by water. In the reaction of Fe(NO) 2(CO)2 with alkali in methanol, [Fe(NO)(CO)3]- is formed, but under comparable conditions Co(NO)(CO)3 gives [Co)CO)4]-, Co(OH)2 and other cobalt-free products. The limited evidence available is consistent with tetrahedral structures for Fe(NO)2(CO)2 (Fig. 5.14) and Co(NO)(CO)3 and a trigonal bipyamidal structure (with NO in the equatorial plane) for Mn(NO)(CO)4 (Fig. 5.15); (Ph3P)2Mn(NO)(CO)2 also has a trigonal bipyramidal structure, the two triphenylphosphine molecules occupying the apical positions. Since Co(NO)3 shows two N-O stretching frequencies in the infrared, it must be pyramidal rather than planar, but the detailed structure is not known. 5.3.5 Nirtosyl Halide Complexes Volatile diamagnetic nitrosyl halides of formula Fe(NO)3X are formed by the action of nitric oxide on iron carbonyl halides in the presence of finely divided iron as a halogen-acceptor. These readily lose NO to give [Fe(NO)2X]2, in which the halogen atoms act as bridges. Analogous compounds of cobalt and nickel may be formed by reactions similar to those involved in the high pressure synthesis of carbonyls; for example, CoX2 + Co + 4NO 2Co(NO)2X 4NiI2 + 2Zn + 8NO 2[Ni(NO)I]4 + 2ZnI2 The ease of formation of these compounds increases in the sequences Ni < Co < Fe and X = Cl < Br < I. Nitrosyl chloride and nickel carbonyl in liquid hydrogen chloride, on the other hand, give Ni(NO) 2Cl2, which is probably monomeric and tetrahedral. Nitrosyl halides are also formed by some metals which, so far as is known, do not form nitrosyls or nitrosyl carbonyls. Thus molybdenum and tungsten (but not chromium) carbonyls react with nitrosyl chloride: M(CO)6 + 2NOCl 20 M(NO)2Cl2 + 6CO CH 2 Cl2 Palladium (II) chloride in methanolic solution yields Pd(NO)2Cl2, and nitrosyl halide molecules or anions are formed also by several other transition metals. 5.3.6 Nirtoso Cyanide Complexes Sodium nitropursside is also a complex resulted from the coordination of NO+. Sodium nitroprusside [nitrosopentacyano-ferrate (II)] is prepared by the action of nitric acid or sodium nitrite on the hexacyanoferrate (II). In the former process the overall reactions is [Fe(CN)6]4- + 4H+ + NO3- [Fe(CN)5NO]2- + CO2 + NH4+ In the latter process, two successive equilibria are involved: [Fe(CN)6]4- + NO2- [Fe(CN)5NO2]4- + CN[Fe(CN)5NO2]4- + H2O [Fe(CN)5NO]2- + 2OHThese are driven to completion by adding barium chloride to the reaction mixture and blowing a current of carbon dioxide through the hot solution to remove the hydrogen cyanide liberated by the reaction 2[Fe(CN)6]4-+2NO2-+3Ba2++3CO2+ H2O 2[Fe(CN)5NO]2-+ 2HCN + 3BaCO3 The formulation of the complex anion as a NO+ derivative of iron (II) is supported by its diamagnetism, a N-O stretching frequency of 1939 Cm-1 and a N-O distance of 1.13A. The purple colour obtained from nitrosopentacyanoferrate (II) and sulphide is [Fe(CN)5(NOS)]4- analogous to [Fe(CN)5NO2]4-. Structure of Nitrosyl Co-ordination Compounds: due to the ion If we compare the electronic structure of NO with CO, it is observed that NO has an additional electron in antibonding π M.O., which may be readily lost to form the nitrosonium ion, NO+. The additional electron present in π molecular orbital of NO can be supplied to metal atom thus increasing its effective number by one unit and neutral No is itself converted into NO+ ion. Then, this NO+ is co-ordinated through nitrogen with the metal atom by donating its lone pair to the metal. 1. Cobalt atom may increase its E.A.N. from 27 to 28 by accepting an additional electron from a neutral molecule of NO: Co + NO Co- + NO+ cobalt ion may then combine with one NO+ group and 3 CO molecules to form stable compounds Co - + NO+ + 3CO Co(NO)(CO)3 In this compound the E.A.N. of Co is, 27+1+2+6 = 36 of stable Krypton. 2. Similarly the formation of Fe(NO)2(CO)2 can be explained. Sidgwick gave the electronic structure of metallic nitrosyls as belowM+ (:N: ::O:+) or M2- - N+ O+ The accumulation of charge on the central atom favours strong π-bond formation with the attached groups. Thus the most of metal nitrosyls are formed by donation from the (NO)+ to the metal atom with the M-O back bonding in a manner analogous to M-C bond in carbonyl it is known as three electron donor M + NO M- + NO+ M2- - N+ = 0+ In terms of M.O.T. the hybrid orbital on N atom having a lone pair [(sp)2N lone pair] overlaps suitable vacant hybrid orbital on M ion (sp3 in tetrahedral or d2sp3 in octahedral) to form ON+ M- bond and the empty π2* or π1* M.O. will overlap with the filled dorbitals to form M- NO+ π bond. This type of overlap transfers some charge from M- ion to NO+ ion. The molecule of NO is a resonance structure of the following forms: π π N O: N – O -N O+: On this basis resonance structures of NO, the metallic nitrosyls may be represented as: π π π π M--N O: M- N O M N O: M--N-O Nitric oxide is a paramagnetic molecule with an electron in an anti-bonding orbital. This electron is relatively easily lost with formation of the NO+ ion and an increase in the N-O stretching frequency from 1878 cm-1 in NO to 2200-2400 cm-1 in nitrosonium salts. Structure of various groups of nitrosyl complexes are shown in Fig. 5.13 to 5.20 Fig. 5.13 Structure of Fe (NO)4 Fig. 5.14 Structure of Fe(CO)2 (NO)2 Fig. 5.15: Structure of [Mn(NO) (CO)4] Fig. 5.16: Structure of [Fe(NO)2I]2 Fig. 5.17: Structure of Fe(NO)3Cl Fig. 5.18: Structure of [Ni (NO)I]4 Fig. 5.19: Red salt of Diethyl Ester of [FeS2(NO)4]2- Fig. 5.20: Anion of Red salt of [Fe4(NO)7S3] 5.4 DINITROGEN COMPLEXES In 1965, Allen and Senoff obtained salts containing the [Ru(NH3)5N2]2+ cation by the action of hydrazine hydrate on various compounds of tri- and tetrapositive ruthenium, amongst them ruthenium trichloride and ammonium hexachlororuthenate (IV). Thus, these substances (often called nitrogenyl or dinitrogen complexes, to distinguish them from those containing the nitride ion) have been known for only a few years. Many other complexes containing one or two (but not, so far, more) molecules of coordinated nitrogen have now been prepared, and it is clear that N2 acts as a -donor and π-acceptor in the same way as isoelectromic CO, though the complexes formed are much less stable than carbonyls. Much of the interest in this field centres on the possibility of developing new methods for nitrogen fixation; up to the present time, however, no method has been found for the reduction of nitrogen in the complexes described here (though this has been achieved by systems involving an organ titanium complex under powerfully reducing conditions). Most, though not all, nitrogenyl complexes have triphenylphosphine and halide or hydride as other ligands in the complex. The following examples illustrate methods for their preparation. (a) The action of nitrogen on a metal complex: for example, CoCl2 + Ph3P NaBH 4 N (Ph3P)3CoH3 (Ph3P)3CoH(N2) EtOH 3 N [Ru(NH3)5H2O]2+ [Ru(NH3)5(N2)]2+ 3 (b) Another method of preparation of dinitrogen complex is the reaction of coordinated azide: [Ru(NH3)5Cl]2+ N3- MeSO3 H [Ru(NH3)5N2]2+ NH 3 (c) Similarly reaction of (Ph3P)2Ir(CO)Cl with RCON3: (Ph3P)2Ir(CO)Cl + RCON3 Ir(PPh3)2(CO)(Cl)(N2.NCOR) CHCl3 / EtOH (Ph3P)2IrCl(N2) (d) Reaction of coordinated NH3 with HNO2: [Os(NH3)5(N2)]2+ + HNO2 [Os(NH3)4(N2)2]2+ + 2H2O The most stable dinitrogen complexes are those of heavier members of iron and cobalt groups. Some are unaffected by dry air and can be heated to 100-200oC without decomposition. Most are rapidly oxidised by air and decompose on heating gently. The orange solid (Ph3P)3CoH(N2+) shows reversible displacement with hydrogen, ethylene or ammonia. Some of the reactions of (Ph3P)2IrCl(N2) (yellow solid) are as follows: (Ph3P)2IrCl(N2) + Ph3P (Ph3P)3IrCl + N2 (Ph3P)2IrCl(N2) + HCl (Ph3P)3IrHCl2 + N2 (Ph3P)2IrCl(N2) + CO (Ph3P)2Ir(CO)Cl + N2 Dinitrogen complexes show an asymmetric IR N N stretching frequency in the range 2230-1920 Cm-1(Raman stretching frequency in N2 is 2331 cm-1). In metal complexes dinitrogen either has a terminal position or as a bridge: N M–N–N N M–N–N–N M M N Terminal Structures of the M N Bridging two important dinitrogen complexes [Ru(NH3)5N2Ru(NH3)5]4+ and [Sm(N5C5Me5)2I2(N2)] are shown in Fig. 5.21 and 5.22 respectively. Fig. 5.21: Structure of [Ru(NH3)5(N2)Ru(NH3)5] Fig. 5.22: Structure of [Sm(N5C5Me5)2I2(N2)] 5.4.1 Fixation of Nirtrogen Dinatrogen complexes while show possibility of developing new methods for nitrogen fixation, they also help in the understanding of the probable mechanism of biological fixation of nitrogen. An important enzyme-system is related with the atmospheric fixation of nitrogen; which involves an important step in nitrogen-cycle and is responsible for supply of nitrogen to the plants growth (e.g. Blue-green algae, symbiotic bacteria legume) The active enzyme in fixation nitrogen is nitrogenase. In this enzyme two proteins take part in the reaction. Small protein has molecular weight of 57000-73000 and contains Fe4S4 group; while the large protein is a tetramer of molecular weight 220000-240000. It has 2 molybdenum atoms, nearly 30 iron atoms and nearly 30 mobile sulphide ions. Fe-S group probably functions as redox centre, and the active site for dinitrogen binding is probably molybdenum atom. (Fig. 5.23) Fig. 5.23: Fixation of Nitrogen 5.5 DIOXYGEN COMPLEXES Amongst all the donor atoms oxygen is most important. The donor ability of oxygen is related with its partial charge; higher is the negative charge, higher will be the donar ability. Large number of coordination compounds are available in which oxygen uses one of its two lone pairs of electrons. The most important example of dioxygen complexation is transportation of oxygen in aerobic-organisms through heme and hemocynin mechanism. Although, hemoglobin and hemocynin are known since long time for their specific ability of absorption and release of oxygen; but now a number of synthetic compounds have this property, e.g. Bis (Salicylic) ethylenedimmine cobalt (II). Heme, protein is the most important group of metallic porphyrin, which functions as a oxygen carrier in aerobic organism. In the centre of its porphyrin ring is iron (Fe2+), which is linked with the protein part of haemoglobin. Heme is very much sensitive for reaction with oxygen and the reactive oxygen complex, forming an intermediate product, is converted into Fe(II) porphyrin or Hemin. As has been shown earlier, heme protein functions as the oxygen carrier during respiration of aerobic organisms. In this process, vertebrates use two heme-proteirs: hemoglobin and myoglobin. Hemoglobin takes dioxygen from lungs or gills and passes it to the tissues. Where it is stored in myoglobin. The cytochromes present in tissues, which functions as electron carrier, reacts with dioxygen and reduces it. The oxidation power of dioxygen is thus used in burring of the food. In this way during transportation storing and use of dioxygen three heme proteins play important part; these are hemoglobin, myoglobin and cytochrome. 5.5.2 Hemoglobin Hemoglobin is the red pigment of blood. It has two parts: (a) 96% part of it is a simple, specific protein called globin and (b) 4% remaining part is the prosthetic group hence: Globin Hemoglobin Heme Protoporphyrin Fe(II) It is a globular protein, which is made up of polypeptide chins. These chains are arranged in a regular tetrahedral form and are linked with the four rings of pyrole. Molecular weight of hemoglobin is nearly 64500. Hemoglobin molecule can coordinate with dioxygen without oxidation of iron. The bonding of iron with dioxygen is so strong that oxyhemoglobin does not decompose during its transportation in the body. Still it is so weak that its contact with oxidase decomposes it readily. The various steps during oxidation of hemoglobin are: Ist step: Bonding with dioxyen: IInd step: Bonded dioxygen links with other heme ( -peroxo complex is formed) : IIIrd step: Decomposition of per oxo complex into ferryl complex. IVth step: Reaction of ferryl Complex with heme to give Hematin: In living being steps I and IV do not take place, otherwise total heme would have precipitated as hematin. Apart from other reactions, steps III and IV are checked by sterric hinderance. Thus dioxygen is carried away by oxyhemoglobin and either stored in oxymyoglobin or given to cytochromes for use. In lungs or gills of vertebrates, the following reactions take place: Hb + 4O2 Hb (O2)4 Hemoglobin Oxyhemoglobin While in tissues, the reaction that takes place is: Hb(O2)4 + 4Mb 4Mb(O2) + Hb Myoglobin 5.6 TERTIARY PHOSPHINE AS LIGAND Large number of triphenylphosphine and similar substituted metal carbonyls are known, e.g. Ni(CO).(Ph3P)2. This compound is of great importance as a catalyst for the polymerisation of olefins and acetylenes e.g. butadiene to cyclooctadiene and acetylene to benzene and styrene. Analogous compounds can be obtained by the action of triphenyl phosphine on iron pentacarbonyl. Similarly dicobalt octacarbonyl gives two products with Ph3P in 1:1 ratio of Co and Ph3P. One compound is [Co2(CO)6(PPh3)2] and the other is the salt [Co(CO)3(PPh3)2][Co(CO)4] in which the cation has the expected trigonal pyramidal structure. A platinum complex, Pt (CO)2(PPh3)2 can be obtained by the action of CO on Pt(PPh3)4. As a matter of fact, substitution of triphenylphosphine for some of the carbonyl groups greatly enhances the stability of the compound; thus although Co(CO)4 I is unstable, (Ph3P)Co(CO)3I can be made by the remarkable reaction. 2 CF I (Ph3P)Co(CO)3I + I- + C2F6 [(Ph3P)Co(CO)3]- 3 Triphenylphosphine carbonyl halides of rhodium and iridium may be prepared by interaction of the metal halide (or a complex halide) and triphenylphosphine in a variety of organic solvents, the solvent serving as the source of the carbonyl group: Ph P (Ph3P)2Ir(CO)CI (NH4)2IrCl6 3 Ph P (Ph3P)2Ir(CO)CI IrCl3.3H2O 3 The product of these reactions- (Vaska's compound) is a highly reactive complex. Vaska's compound is a carbonyl halide; and many triphenylphosphine complexes containing rhodium and iridium show similar reactivity and catalytic activity. The iridium compounds is remarkable for its reversible uptake of H 2, O2 and SO2 to give crystalline 1:1 adducts which can be decomposed by lowering the pressure; for example, O (Ph3P)2Ir(CO)Cl O2Ir(PPh3)2(CO)Cl 2 In the oxygen adduct, oxygen atoms occupy cis octahedral positions; the O-O distance of 1.30 Ao suggests that Oxygen is present as O2- rather than O22-. Some of the many other reactions of Vaska's compounds are shown in Fig. 5.24 Fig. 5.24 Check Your Progress-2 Notes :(i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. A.(i) Most of the metal nitrosyls are formed with..................ion. However, most of the pure nitrosyl complexes have the general formula............... (M = ...........................................) (ii) The various modes of linking of NO are: (a) ...................................... (b) ...................................... (c) ...................................... (d) ...................................... B. Fixation of nitrogen involves enzyme..........................., which has two proteins. The small contains........................group; protein, while mol-weight....................., the large protein, mole weight......................... contains................Mo atoms...................Fe atoms and............mobile sulphide ions. C. (i) Hemoglobin binds dioxygen to give..........................: (reaction)................................................................................ (ii) The product is given to...............................for storage and (iii) is used by...........................for burning of food. D. Vaska compound has general formula.................................... 5.7 LET US SUM UP Most transition metals form complexes with a wide variety of unsaturated molecules, such as CO, NO, O2, N2 etc., using ML π-bonding, which stabilize these complexes. CO molecules combine with transition metal atoms (generally in zero oxidation state) to give series of carbonyls, varying from mononuclear, di-nuclear, trinuclear, tetra-nuclear to hexa-nuclear carbonyls. In which EAN rule is strictly followed. Metals with even number of electrons give stable mononuclear carbonyls; but the metals possessing odd number of electrons do not form stable mononuclear carbonyls. The shortage of one electron is compensated by linking with H or Cl or by dimmer formation, e.g. V(CO)6 forms H[V(CO)6, Na[V(CO)6], [V(CO)6]Cl or V2(CO)12. IR spectra give important information regarding the nature of CO group in the complex. Thus the terminal CO group indicate by the stretching frequency at 21251850cm-1 (or 2125-2000cm-1). While the bridging COgroup is indicated by the stretching frequency at 19001800 cm-1 region. Frequency at 1883 and 1788cm-1 respectively are indicative of strong π-bonding (ML). Ni(CO)4 is tetrahedral, Fe(CO)5 is TBP, Cr(CO)6 is octahedral while the di-nuclear, trinuclear, tetranuclear and hexanuclear carbonyls have structures derived from linking of octahedral in respective numbers of sharing corners or side or a face. Nitric oxides combine with transition metals to form coordination compounds. The general modes of linking may be- (a) (b) (c) (d) D :N D :N :N :N :O: Links with bonds (neutral) :O: :O: :O: Links with Links with Links with and bonds dative bonds dative bonds (cationic) (cationic) (cationic) Most of the nitrosyl complexes are derived from linking of NO+ (nitrosonium ion). Pure nitrosyls have general formula M(NO)4 with M = Fe, CO, Ru. However Co(NO)3 has also been reported. NO displaces CO from V(CO)6, (Ph3P)2Mn(CO)8, Fe2(CO)9 and Mn(NO)(CO)4, Co2(CO)8 to Fe(NO)2(CO)2 give V(NO)(CO)5, and Co(NO)(CO)3 nitrosylcarbonyl complexes respectively. In addition to these, binuclear species such as Mn2(NO)2(CO)7 are also formed. Many dinitrogen complexes have been reported e.g. [Ru(NH3)5N2], [(NH3)5RuN2Ru(NH3)5], [(Ph3P)2IrCl(N2)], [Os(NH3)4(N2)2] etc. Dinitrogen complexes while show possibility of developing new methods for nitrogen fixation, they also help in the understanding of the probable mechanism of biological fixation of nitrogen. The active enzyme in fixation of nitrogen is nitrogenase. The enzyme has two proteins one small (mol wt. 5700073000) protein contains Fe4S4 groups; while the large protein (mol wt. 220000-240000) is a 2 2 tetramer, which has 2 Mo atoms, nearly 30 Fe atoms and nearly 30 mobile sulphide ions. Fe-S group functions as a redox centre and the active site for dinitrogen binding is molybdenum atom. Amongst all the donar atoms oxygen is most important. This ability is related with its partial charge. The most important example of dioxygen complexation is transportation of oxygen in aerobic organisms, through heme and hemocynin mechanism. During respiration of aerobic organisms two heme proteins, hemoglobin and myoglobin, are used. Hemoglobin takes dioxygen from lungs or gills and passes it to the tissue where it is stored in myoglobin. The cytochromes present in tissues use the oxidation power of dioxygen in burring of food. Hb + 4O2 Hb(O2)4 Hemoglobin Oxyhemoglobin Hb(O2)4 + 4Mb 4Mb(O2) + Hb Myglobin Oxyhemoglobin Large numbers of triphenyl phosphine and similar substituted metal carbonyls are known e.g. Ni(CO)(Ph3P)2. This compound is of great important as a catalyst for polymerisation of olefins and acetylenes. Substitution of triphenyl phosphine for some of the carbonyl groups greatly enhances the stability of the compound. Most widely studied compound is 'Vaska compound' (Ph3P)2Ir(CO)Cl, which is used for the preparation of large number of triphenyl phosphine containing complexes. 5.8 CHECK YOUR PROGRESS: THE KEY 1.(a) Involve CO M and M CO -bonding. (b) Form mononuclear carbonyl....................give dimers or mononuclear carbonyls linked with H or Cl. (c) At 2125-2000 cm-1 and at 1900-1800 cm-1 at 1883 and 1788 cm-1 respectively (d) Has no bridging group, Fe2(CO)9 has three bridging groups. 2(A) (i) With NO+ ion formula M(NO)4 (M = Fe, CO and Ru). (ii) (a) (b) :N :N :O: Links with bonds (neutral) :O: Links with and bonds (cationic) (c) D :N (d) D :N :O: :O: Links with Links with dative bonds dative bonds (cationic) (cationic) B. Enzyme nitrogenase, small protein mol. wt 57000-73000 contains Fe4S4 group Large protein mol. wt. 220000-240000 contains 2 Mo atoms 30 Fe atoms and 30 mobile sulphide ions. C.(i) To give Oxohemoglobin Hb + 4O2 Hb(O2)4 (ii) Myoglobin for stroage and (iii) by cytochromes D. Vaska compound has general formula: (Ph3P)2Ir(CO)Cl Unit - 6 REACTION MECHANISM OF TRANSITION METAL COMPLEXES-I Structure 6.0 Introduction. 6.1 Objectives. 6.2 Energy Profile of a Reaction. 6.3 6.2.1 Reactivity of metal Complex - Inert and Labile Complexes. 6.2.2 Valence Bond and Crystal Field applications. Kinetics of Octahedral Substitution 6.3.1 Nucleophilic Substitution 6.3.2 Hydrolysis Reactions 6.3.3 Factors affecting Acid Hydrolysis 6.3.4 Base- Hydrolysis-Conjugate Base Mechanism 6.3.5 Anation Reaction 6.3.6 Reactions without Metal-Ligand Bond-Cleavage 6.4 Let Us Sum Up 6.5 Check Your Progress: The Key 6.0 INTRODUCTION Metal complexes are generally classified as 'Labile" and 'Inert' with reference to their reactivity. The ability of a complex to engage itself in reactions involving the replacement of one or more ligands in its coordination sphere by other ligand is called lability of the complex. The complexes that undergo rapid substitution are termed labile. Where as those with low rates of substitution are called inert. However, the degree of lability or inertness of a transition metal complex can be correlated with the delectron configuration of the metal ion. Nearly half of all reactions of transition metal complexes may be considered substitution reactions, while the remaining half are redox-reactions. Equilibrium and kinetics play important and central part, for determining the outcome of inorganic reactions. It is often helpful to understand the mechanism of the reactions. A chemist who wishes to synthesise an octahedral complex, as an example, must have some idea of lability of the complex in order to choose appropriate experimental conditions for synthesis. Because the mechanism is rarely known finally and completely, the nature of the evidence for a mechanism should always be kept in mind in order to recognise what other possibilities might also be consistent with it. In the first part of this unit we describe how reaction mechanisms are classified, and distinguish between the steps by which the reaction takes place and the details of the formation of the activated complex. Then these concepts are used to describe the currently accepted mechanisms for the substitution reactions of complexes. However, you may recall what you have already studied about the basic concept of kinetics and of current views on the nature of substitution at a saturated carbon atom, as inevitably organic chemical thinking has expected a great influence on the interpretation of the kinetics of inorganic reactions. 6.1 OBJECTIVES The main aim of this unit is to look in detail at the evidence and experiments that are used in the analysis of reaction pathways and develop a deeper understanding of the mechanism of substitution reactions of d-block metal complexes. After going through this unit you should be able to: discuss the energy profile of a reaction and explain lability in terms of VBT and CFT principles; describe nuelcophilic substitution reactions in octahedral complexes in terms of SN1 and SN2 reaction mechanisms, and the evidences supporting them; explain acid- and base-hydrolysis reactions and their mechanisms; explain water exchange (Anation) is a binuclear reaction and the rate of this reaction depends upon the nature of the metal ion; and 6.2 describe the catalysts form octahedral substitution reactions; ENERGY PROFILE OF A REACTION. Why does a chemical reaction take place? What happens in a chemical reaction? Answer of these and similar other questions are important for a chemist; so that he can have control over a chemical reaction and can either complete it or stop it, according to the need. In order to convert reactants into product, it is necessary that the groups or the atoms, linked in what ever manner, in the reactant molecules should separate (may be partially) and then reunite (re-link) in the form of the products. Unless this takes place using a suitable mechanism, the reaction will not take place. On the thermodynamic basis, the possibility of conversion of reactants, into products is only when the state of disorder and the bond-energies in the products are relatively high. Both of these, affect the direction of a chemical change and on the effect of these depends the important thermodynamic functions Gibb's free energy, G. For a chemical reaction, free energy is related with the heat content or the useful energy, H , and the disorder, S , according to the following relation: G = H - T S That is, a chemical reaction will go in the direction in which there is decrease in free energy, i.e. G should be negative. In order to a chemical reaction takes place, (i) the total bond energies in the product are stronger then that in the reactants and the total disorder (entropy) of the products is high or (ii) the total bond forces in the products are stronger than that in the reactants and the products is less, but T S is greater than H or (iii) the total bond forces in the products are weaker as compared to that in the reactants, but the increase in entropy is so high that it compensates the energy absorbed. 6.2.1 Reactivity of Metal Complex - Inert And Labile Complexes Almost all the reactions of transition metal complexes may be divided into two categories; (a) Substitution reactions and (b) Redox-reactions. In coordination chemistry rate of a reaction is equally important, as the reaction equilibrium. The ability of a complex to engage itself in reactions involving the replacement of one or more ligands in its coordination sphere by other ligands is called the lability of the complex. The complexes that undergo rapid substitution (half time period or T1/2 or reaction rate k is used to denote the speed of the reactions) are termed labile, whereas those with low rates of substitution are called inert. The inertness of the complex has nothing to do with the stability as determined thermodynamically. Thus, [Ni(CN)4]2-,[Mn(CN)6]3- and [Cr(CN)6]3- all have high stability constants. Yet the rate of exchange of CNby the labelled 14 CN- gives half time period as 30 S, 1 h and 24 days, respectively. Therefore [Ni(CN)4]2- is labile, while [Cr(CN)6]3- is inert. Similarly [Fe(H2O)6]3+ (bond energy = 690 KJ mol-1) is labile while [Cr(H2O)6]3+ is inert. [Ni(CN)4]2- is thermodynamically stable but kinetically labile but [Co(NH3)6]3+ is kinetically inert but thermodynamically unstable. 6.2.2 Valence Bond (VBT) And Crystal Field (CFT) Applications (a) VBT Application According to VBT Octahedral Complex are of two types: i. Outer-Orbital Complexes which involve sp3d2 hybridisation. ii. Inner-Orbital Complexes which involve d2sp3 hybridisation. The two d-orbitals involved in sp3d2 and d2sp3 hybridisation are dx2-y2 and dz2 eg set orbitals. 1. Outer-Orbital Octahedral Complexes Outer-orbital octahedral complexes (sp3d2 hybridisation) are generally labile for example the octahedral complexes of Mn 2+(3d5)Fe2+, Fe3+(3d5) Co2+ (3d7) Ni2+ (3d8) Cu2+ (3d9) and Cr2+ (3d4) exchange ligands rapidly and hence are labile. This is because, the use of outer d-orbitals does not make effective overlap between metal and ligand orbitals resulting in weaker bonds. 2. Inner-Orbital Octahedral Complexes The six d2sp3 hybrid orbitals are filled with the six electron pairs denoted by the 6 ligands. dn electrons of the central metal will occupy dxy, dyz and dxz orbitals. These complexes are inert as the use of inner d-orbitals results in an effective overlap between metal and ligand orbitals giving stronger bonds. Inner orbital octahedral complexes are given in the Table 6.1 which explain the following observations: a. In the labile inner-orbital octahedral complexes there is at least one d-orbital of t2g set empty, so that this empty d-orbital may be used to accept the electron pair from the incoming ligand in forming the transition state with coordination number seven(unstable), which finally stabilise in to an octahedral complex (Coordn. No. 6), removing one ligand (Fig 6.1). Fig. 6.1 b. In the inert-orbital octahedral complexes every d-orbital of t2g set (i.e. dxy, dyz and dxz) contains at least one electron, and have no vacant orbtial to link an extra ligand. Table 6.1 Distribution of dn-electrons in various t2g orbitals for labile and inert inner-orbital octahedral complexes (according to VBT) n n Type of the d Distribution of d electron (shown by Example of central complex configu arrows) in t2g orbitals. Electrons shown by metal ions ration crosses in eg orbitals have been donated by six ligands to enter d2sp3 hybrids and are in opposite spins. d s p t2g eg 2 2 xy yz zx x -y z2 px py pz 0 inner d xx xx xx xx xx xx Sc(+3), Y(+3), rare orbital earth (+3), Te(+4), labile Zr(+4), Hf(+4), Ce(+4), octahedral Th(+4), Nb(+5), Ta(+5), complexes Mo(+6), W(+6) d1 xx xx xx xx xx xx Ti(+3), V(+4), Mo (+5), W Re(+6) d2 xx xx xx xx xx xx Ti(+2), V(+3), Nb (+3), Ta(+3), W(+4), Re(+5), Ru(+6) inner orbital inert octahedral complexes d3 xx xx xx xx xx xx V(+2), Cr(+3), Mo(+3), W(+3), Mn(+4), Re(+4) d4 xx xx xx xx xx xx [Cr(CN)6]4-, Mn(CN)6]1 Re(+3), Os(+3), Ir(+4) Type of the dn Distribution of dn electron (shown by complex configu arrows) in t2g orbitals. Electrons shown by ration crosses in eg orbitals have been donated by six ligands to enter d2sp3 hybrids and are in opposite spins. d s p t2g eg 2 2 xy yz zx x -y z2 px py pz d5 xx Example of central metal ions xx xx xx xx xx [Mn(CN)6]4-, Re(+2), Fe(CN)6]3- Ru(+3), Os(+3), Ir(+4) d6 xx xx xx xx xx xx [Fe(CN)6]4-, Ru(+2), Os(+2), Co(+3) (except Co Fe34Rh (+3), Ir (+3) Table 6.2 Loss in CFSE, E (in the units of Dq) in the formation of a pentagonal bipyramidal intermediate in octahedral substitution reactions on the basis of SN2 associated mechanism SN2 association mechanism Octahedral (oct.)Pentagonal bipyramidal (pent.bipy.) (C.N. = 6) dn ion (C.N. = 7) Strong ligand field (spin-paired or Weak ligand field (spin-paired or low-spin complexes) low-spin complexes) Oct. pent.bipy. (C.N.= 6) (C.N.= 7) d0 0 Dq 0 Dq d1 4 5.28 E E Oct. pent.bipy. (C.N.= 6) (C.N.= 7) 0 Dq 0 Dq 0 Dq 0 Dq 0 4 5.28 0 SN2 association mechanism Octahedral (oct.)Pentagonal bipyramidal (pent.bipy.) (C.N. = 6) dn ion (C.N. = 7) Strong ligand field (spin-paired or Weak ligand field (spin-paired or low-spin complexes) low-spin complexes) Oct. pent.bipy. (C.N.= 6) (C.N.= 7) d2 8 10.56 d3 12 d4 E E Oct. pent.bipy. (C.N.= 6) (C.N.= 7) 0 8 10.56 0 17.74 -4.26 12 7.74 -4.26 16 13.02 -2.98 6 4.93 -2.07 d5 20 18.30 -1.70 0 00 0 d6 24 15.48 -8.52 4 5.28 0 d7 18 12.66 -5.34 8 10.56 0 d8 12 7.74 -4.26 12 7.74 -4.26 d9 6 4.93 -1.07 6 4.93 -1.07 d10 0 0.00 0 0 0.00 0 Table 6.3 Loss in CFSE, E (in the units of Dq) in the formation of a pentagonal bipyramidal intermediate in octahedral substitution reactions on the basis of SN1 associated mechanism dn ion SN1 association mechanism Octahedral (oct.) Syware Pyramidal (Squ. pyi) (C.N. = 6) (C.N. = 5) Strong ligand field (spin-paired Weak ligand field (spin-paired or low-spin complexes) or low-spin complexes) d0 Oct. (C.N.= 6) 0 Dq pent.bipy. (C.N.=5) 0 Dq d1 4 d2 E E 0 Dq Oct. (C.N.= 6) 0 Dq pent.bipy. (C.N.= 5) 0 Dq 0 Dq 4.57 0 4 4.57 0 8 9.14 0 8 9.14 0 d3 12 10.00 -2-00 12 10.00 -2 d4 16 14.57 -1.43 6 9.14 0 d5 20 19.14 -0.86 0 00 0 d6 24 20.00 -4.00 4 4.57 0 d7 18 19.14 0 8 19.14 0 d8 12 10.00 -2.00 12 10.00 -2 d9 6 9.14 0 6 9.14 0 d10 0 0.00 0 0 0.00 0 The value of CFSE mentioned are in the units of Dq and have been given for both the fields viz. strong field and weak field and for both the mechanism (SN1, and SN2). Negative values of E denotes a loss of CFSE when octahedral complex is changed into an activated complex which may be square pyramidal or pentagonal bipyramidal. If the CFSE of the activated complex is greater than that of octahedral complex. E has been given zero value which shows that these complexes do not loose CFSE when they are changed into activated complexes. The octahedral complexes formed by the ions for which there is large loss in CFSE are least labile i.e. such complexes are inert. On the other hand octahedral complexes given by ions for which there is little or no loss in CFSE are labile i.e. such complexes react rapidly. Thus we see: i Both high spin and low spin octahedral complexes of d 0, d1 and d2 ions will react rapidly, i.e. these are labile complexes, in which there is no loss in CFSE. ii. According to VBT inner-orbital octahedral complexes of d3, d4, d5, and d6 ion are inert while these are called low spin or spin paired complexes according to CFT. CFT predicts that low spin complexes of these ions are also inert whether the mechanism is assumed to be SN1 or SN2 in which CFSE values decreases. The ion with maximum loss of CFSE will form the most inert complex. Thus the order of inertness of low spin complexes formed by d 3, d4, d5 and d6 ions is: Order of inertness : d6 > d6 > d4 > d5 Loss of CFSE for SN1 mechanism : -4.00>-2.00>-1.43>-0.86 Loss of CFSE for SN2 mechanism : -8.52-4.26-2.98-1.70 The order of reactivity will be reverse of the above i.e. the order of reactivity will be d6 > d3> d4 > d5 it is supported by the following facts: i. High spin octahedral complexes of d3 ion will react slowly, i.e. these are inert complexes because for this ion there is substantial loss in CFSE whether the substitution mechanism is SN1 or SN2. ii. High spin octahedral complexes of d5 ion react rapidly i.e. these are labile complexes, since there is no loss in CFSE. iii. Both high spin and low spin octahedral complexes of d8 ion are inert. According to VBT d8 ion [3dxy2, 3yz2, 3dxz2, 3d(x2-y2), 3dz2] will form outer orbital complexes and will be labile. Therefore in case d8 ion VBT & CFT gives different predictions. iv. Both high spin and low spin octahedral complexes of d10 ion are labile. Factors Affecting the Liability of Complex 1. Charge of the metal ion: For the isoelectronics complexes there is a decrease in lability with the increase of the charge of the central metal ion. i. The order of lability of the complex is as follows: Lability order : [AlF6]3- > [SiF6]2 > [PF6]- > [SF6]0 Cationic charge : +3 < +4 < +5 < +6 ii. The rate of water exchange represented by: [M(H2O)6]n + 6H2O* [M(H2O*)6]n+ + 6H2O decreases with the increase of cationic charge in the series Rate of water exchange: [Na(H2O)6]+ > [Mg(H2O)n]2+ > [Al(H2O)6]3+ Cationic Charge +1 < +2 < +3 2. Radii of the Central ion : Complexes having central atoms with small ionic radii react more slowly than those having larger central ions i.e. the lability increase with the increase of ionic radius e.g. Order of liability: [Mg(H2O)6]2+<[Ca(H2O)6]2+< [Sr(H2O)6]2+ Cationic Size (A) 0.65 < 0.99 < 1.13 3. Charge to Radius Ratio Values: Octahedral complexes having the central metal ion with the largest charges to radius ratio will react slowest (Fig. 6.2). i. The first row transition elements [Ni(H2O)6]2+ (a d8 system) has the largest value of half life i.e. it reacts slowest. The hydrated M2+ ions [M(H2O)x]2+ of the first row transition elements are all high spin complexes. ii. [Cu(H2O)6]2+ reacts most rapidly because the 2 water molecules above and below the square plane of the tetragonal distorted octahedral shape of [Cu(H2O)6]2+ are exchanged. The remaining four H2O molecules lying in the square plane react slowly. Fig. 6.2: Half-lives (in sec) at 25oC for the exchange of water by some hydrated metal ions. 4. Geometry of the Complex: Four co-ordinated complexes react more rapidly than analogues 6-co-ordinated complexes e.g. the very stable [Ni(CN)4]2+ undergoes rapid exchange with 14CN-, [Ni(CN)4]2+ + 414CN- [Ni(14CH)4]2- + 4CN while 6-co-ordinated complexes like [Mn(CN)6]4- and [Co(CN)6]3have the same stability as [Ni(CN)4]2+. The greater reactivity of 4co-ordinated complexes may be due to the fact there is enough room round the central ion for the entry of a 5th group into the coordination sphere to form on activated complex. Check Your Progress-1 Notes : (i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. A.(i)For a reaction to go in the forward direction G should be................................ (ii)According to the thermodynamic relation G = ......................... That is for conversion of the reactants into the products, the bond energies and the state of disorder should be....................i.e. the value of the H should be.....................and that of T S should be............................... B.(i) According to VBT, generally labile complexes are........................ complexes, while the inert complexes are.................complexes. (ii) Inner orbital complexes may be labile, if they have at least.............. in.........................set is vacant, e.g. in.................. (iii) According to CFT inert complexes have............................values of ................................ 6.3 KINETICS OF OCTAHEDRAL SUBSTITUTION Substitution reactions involve the activated complex which is most unstable changes to give the product x-y and z. Thus the various steps responsible for the reaction are X + Y - Z X.......... Y..........Z X - Y + Z Reactants Activated complex (Transition State) Unstable Products The difference in energy between the reactants and the activated complex is called activation energy. These reaction involves two process (1) SN1 and SN2 1. In SN1 process the rate-determining slow step is a metal-ligand bond breaking step, since the co-ordination No. of the complex MX5Y (=6) is decreased to 5 which is the co-ordination number of the intermediate MX5. For a ligand replacement reaction of the general type [LnMX] + Y = [LnMY] + X (For simplicity all charges are omitted), the mechanism analogous to unimolecular nucleophilic substitution (sN1) at a carbon atom would be: slow [LnM] + X [LnMX] fast [LnMY] + X [LnM] + y The rate of SN1 mechanism is first order with respect to MX5Y, i.e. the rate-determining step in this mechanism is unimolecular. On the other hand the rate determining step for SN2 mechanism is bimolecular, i.e. its rate of reaction is second order first order with respect to MX5Y and first order with respect to Z. Thus for SN1 mechanism rate = K [MX5Y], and for SN2 mechanism rate = K [MX5Y][Z] Here, it may be mentioned that the kinetic data would be equally compatible with ion-pair formation (if both reactants are ions) followed by a unimolecular reaction of the ion-pair: k1 [Ln MX] + Y [Ln MX] Y slow [Ln MY] + X [Ln MX] y This leads to k k [ L MX ][Y ] d [Ln MY] = 1 2 n dt k 1 k 2 = k[Ln MX][Y] where k= k1k 2 k 1 k 2 Detailed investigation of such a reaction can lead to a value for k 1/k-1, the equilibrium constant for ion-pair formation. 6.3.1 Nucleophilic Substitution As has been motioned, nucleophile substitution reactions in octahedral complexes follow either of the two mechanisms, the dissociation mechanism or the SN-1 mechanism and the association mechanism or the SN-2 mechanisms. The rate determining step in association or dissociation, may be worked out by analysing the rate-laws of the reactions taking place and the specific conditions under which the reactions take place. The difference in these two mechanisms depends on, whether the rate determining steps is the formation of a new Y...............M bond or the dissociation of an old M...................X bond. (a) SN-1 or Dissociation Mechanism The nucleophilic substitution unimolecular reaction actually proceeds in two steps. In the first, slow and rate determining step, one ligand Y is lost and a five coordinated intermediate is formed. In the second step the short-lived penta-coordinated intermediate of very limited stability is attacked rapidly by the nucleophilic reagent, Z to give the complex, MX5Z. There two steps are diagrammatically shown in Fig. 6.3 Fig. 6.3: SN1 or dissociation mechanism for the substitution reaction MX5Y + Z MX5Z + Y For the SN1 mechanism, the following points are important: (1) The trans effect of the ligands would not be operative due to the dissociation of the ligand completely from the octahedral complex. (2) The rates of SN1 substitution (k1) should be inversely proportional to the strength of the Co-L bond, and depend on the charge, steric factors, and chelating effects of the leaving group L. (3) Increase in the electron density on Co atom by the electron donors in SNn should assist the M-L bond breaking. (4) k1 is independent of the nature of E as well as its concentration except for the OH- group for which the reaction is of the second order. (5) Cis effect. Ligands having another pair of electrons like CNS - or OH- increase the rate of hydrolysis of the complexes about ten fold when present cis to L, as compared to the rate when they are present trans to L. This is due to the stabilization of the square pyramidal complex by the electron pair donation by OH- or CNS- along the Cis position through p-d- bonding (Fig. 6.4). No rearrangement takes place and the product is 100 percent Cis isomer. The ligands that do not show the eis effects are those that do not have an extra pair of electrons (NH3) or are themselves acceptors (NO2-, CO, NO, etc.). Fig. 6.4: Cis-effect From Table 6.4, it can be seen that for the formation of the 5coordinate intermediate, high energy changes are required for the low spin d3, d6 and d8 and high spin d3 and d8 ions. Hence, these complexes do not favour the SN1 mechanism for the substitution. Table 6.4 Changes in LFSE (in Dq) for Changing a 6coordinate Complex to a 5-Coordinate (SP) or a 7-Coordinate (Pentagonal Bipyramid) species. System do, d10 High spin CN = 5 CN = 7 0.00 0.00 Low spin CN = 5 CN = 7 0.00 0.00 d1 0.57 1.28 0.57 1.28 d2 1.14 2.56 1.14 2.56 d3 -2.00 -4.26 -2.00 -4.26 d4 3.14 -1.07 -1.43 -2.98 d5 0 0 -0.86 -1.70 d6 0.57 1.28 -4.00 -3.52 d7 1.14 2.56 1.14 -5.34 d8 -2.00 -4.26 -2.00 -4.26 d9 3.14 -1.07 3.14 -1.07 + value indicate gain in CFSE while - values indicate loss in CFSE. (b) SN-2 or Association Mechanism SN-2 or the nucleophilic bimolecular substitution reaction also proceeds through two steps: The first step is slow step and involves the attachment of the incoming nuclepohile, Z to MX5Y to form a seven-coordinate unstable intermediate (perhaps transition state) which is probably pentogonal bipyramidal in shape. Obviously it is a metal-ligand bond-making step. ( Z ) MX5Y Slow (C.N.=6) MX5YZ Unstable seven-coordinatee Intermediate (C.N.=7) This reaction is rate-determining and bimolecular because two reactants viz MX5Y and Z are involved in this step. Thus the rate of this rate-determining reaction is of second order: first order with respect to the complex, MX5Y and first order with respect to the entering ligand, Z, i.e., Rate of reaction = K[MX5Y][Z] In the second step either at the same as Z adds to MX5Y or shortly thereafter, Y leaves MX5YZ rapidly to give MX5Z. This is a fast step. MX5YZ Unstable seven- Fast MX5Z -Y (C.N.=6) coordinatee Intermediate (C.N.=7) Both these steps are shown diagrammatically in Fig. 6.1 This mechanism is similar to Eigen-Wilkins Mechanism, which presents formation of the association complex [L-MX5-Z] in the preequilibrium step: Thus the following equilibrium will be established: LMX5 + Z MX5. Z; K = [ LMX 5 .Z ] [ LMX 5 ][ Z ] The value of the equilibrium constant, K, for the association complex, may be obtained using Fuoss-Eagan equation, K= where, 4 a3 NAe-v/RT 3 a = Nearest reach-distance v = Coulomb potential energy at a-distance NA = Avogadro number = (Z1Z2e2/4 ea) As in the octahedral complexes, the six ligands are already present along the three C4 axes along which the eg orbitals are concentrated, the t2g orbitals that lie along the C2 axes most probably have to be approached by the seventh ligand to form the associated complexes in the SN2 process. Hence, if the t2g orbitals are filled (Co2+ in low spin octahedral complexes), the higher activation energy required to empty one of the t2g orbitals will make the complex inert. Table 6.4 also shows that due to the loss of the CFSE energies, the d3 and low spine d6 ions require highest activation energies, followed by d7, d8 (Ni2+complexes are labile due to the expulsion of ligand by the eg orbitals) and high spin d3 and d8 ions. Thus, SN-1 and SN-2 reactions differ in the following points: (i) In SN1 process the rate-determing slow step is a metal-ligand bond breaking step, since the coordination number of the complex, MX 5Y (=6) is decreased to 5 which is the coordination number of the intermediate, MX5. On the other hand in SN2 process the ratedetermining step involves a metal-ligand bond making step, since C.N.=6 is increased to 7. (ii) The rate of SN1 mechanism is first order with respect to MX5Y, i.e., the rate-determining step in this mechanism is unimolecular. On the other hand the rate-determining step for SN2 mechanism is bimolecular, i.e. its rate of reaction is second order: first order with respect to MX5Y and first order with respect to Z. Thus: for SN1 mechanism rate = K[MX5Y] and for SN2 mechanism rate = K[MX5Y][Z] 6.3.2 Hydrolysis Reactions The substitution reactions in which a ligand is replaced by a H 2O molecule or by OH- groups are called hydrolysis reactions. They are of two types (a) when an aqua complex is formed by the replacement of a ligand by H2O molecules are called acid hydrolysis or equation reactions, while (b) the reactions, in which a hydroxo complex is formed by the replacement of a ligand by OH- group are called base hydrolysis reactions. Acid hydrolysis reactions occur in neutral and acidic solutions (pH <3) while base hydrolysis reactions occur in basic solution (pH 10). Examples are:[CoIII(NH3)5Cl]2+ + H2O[CoIII(NH3)5(H2O)]3+ + Cl- Acid hydrolysis [CoIII(en)2ACl]+ + H2O [CoIII(en)2A(H2O)]2+ + Cl- reaction [A = OH-, Cl-, NC-, NO2-] [Co(NH3)5Cl]2++OH-[Co(NH3)5(OH)]2++Cl- (Base hydrolysis reaction) (a) Acid Hydrolysis or Aquation : When NH3 or ammines like ethylene diamine or its derivatives co-ordinated Co(III) are replaced very slowly by H2O molecules and hence in acid hydrolysis only the replacement of ligands other than amines is usually considered. The rate of hydrolysis of the reaction is of first order. [Co(NH3)5X]2+ + H2O[Co(NH3)5(H2O)]3+ + XThe rate of hydrolysis reaction is of first order. In aqueous solution the concentration of water is always constant, the effect of changes in water concentration on the rate of the reaction cannot be determined. The rate law K = K1[Co(NH3)5X]2+[55.5] does not indicate whether these reactions proceed by an SN2 displacement of X by H2O or by an SN1 dissociation followed by the addition of H2O. The rate law for acid hydrolysis at low pH thus becomes - d [Co(NH3)5X] = kA[Co(NH3)5X] dt (If X is the anion of a weak acid, a term kH+[Co(NH3)5X][H+] is added.) As we have shown previously, such a rate law is compatible with either a slow dissociation of the complex into [Co(NH 3)5]3+ and X or replacement of X by H2O as the rate-determining step. In order to try to decides between these alternatives, the rates of hydrolysis of a series of complexes of formula [Co(AA)2Cl2]+, where AA is a substituted ethylendiamine, were examined. For replacement of a single chloride ion at pH 1 the order found for values of kA was CH2NH3 CH3CHNH2 < CH2NH3 CH3CHNH3 < CH2NH2 (CH3)2CNH2 < CH3CHNH2 (CH3)2 CNH2 Such an acceleration of substitution by bulky ligands suggests that the dissociative mechanism is operative; although introduction of methyl groups must have some inductive effect, the variation in base strengths among the diamines is very much less than the variation in rate constants for the hydrolysis of their cobalt (III) complexes, and it seems reasonable to attribute the kinetic effect mainly to steric factors. Now since steric factors favour SN1 reactions, this is evidence for the dissociative mechanism. Further evidence for this mechanism is provided by: (a) a general inverse correlation between the rate of replacement of X in [Co(NH3)5X] and the formation constant of the [Co(NH3)5X] complex from [Co(NH3)5(H2O)]3+ and X, and (b) the decrease in the rate of the exchange reaction [Co(NH3)5(H2O)]3+ H218O = [Co(NH3)5(H218O)]3+ + H2O at high pressures. 6.3.3 Factors Affecting Acid Hydrolysis (i) Effect of Charge on the Complex: The value of rates of acid hydrolysis of some Co(III) complexes at pH=1 shows that the divalent monochloro complexes react about 100 times slower than the monovalent dichloro complexes. As the charge of the complex increases, a decrease in rate is observed and the acid hydrolysis of the divalent complexes like [Co(NH 3)4(H2O)Cl2]2+ occurs in two steps: [Co(NH3)4 (H2O)Cl]2+ + slow Cl 6-co-ordinate complex [Co(NH3)4(H2O)]3+ + [Co(NH3)4(H2O)]3+ + Cl5-co-ordinate Intermediate fast H 2O [Co(NH3)4(H2O)2]3+ The acid hydrolysis represented by equation (1) would proceed more rapidly than that represented by equation (2) because the separation of a negative charge in the form of Cl ion from a complex ion with higher charge is more difficult. (ii) Effect of Chelation When NH3 molecules in [Co(NH3)5Cl]2+ complex ion are replaced partially or completely by polyamines like en, trien, diene, tetraene etc, the rates of the reaction of the divalent complex ions shows that as the number of -CH2-CH2 or -(CH2)2-chelated links increases the rate values decreases. The replacement of NH3 molecules by polyamines increases the size of the complex i.e. the chelated complex has larger size. The larger the size of the ion less its solvation energy will be and hence less easily it will be formed. Thus the stability of the transition state in which the Cl ion is only partially lost and in which the solvation is less efficient will be reduced. The rate of equation is slowed down by chelation because of reduced stability of the transition state due to less efficient solvation. (iii) Effect of Substitution on ethylene diamine When H atoms on carbon atom or on nitrogen atom of en groups of trans [Co(en)2Cl2]+ are replaced by the alkyl groups like CH3,C2H5 etc. the ligand becomes more bulky, if the strained complex having bulky ligand reacts by SN1, dissociative mechanism and co-ordination number 6 is reduced into 5 co-ordinated intermediate, on the other hand if the strained complex reacts by SN2 displacement process, the crowding on the complex is increased as it is converted into a transition state of co-ordination number seven. The rate of hydrolysis of trans [Co(AA2 Cl2)]+ at 25oC and pH=1 corresponding to the replacement of only one Cl- ion by H2O molecule are given. Here AA is the diamine. (iv) Effect of Leaving Group The rate of reaction of [Co(NH3)5X]2+ corresponding to the replacement of X with H2O molecule depends on the nature of X because the bond breaking step is important in rate determining step. The reactivity of Xgroups decreases in the order. HCO3->NO3->I->Br->Cl->SO4-->F->CH3COO->SCN-<NO2 6.3.4 Base- Hydrolysis-Conjugate Base Mechanism The base hydrolysis reaction represented by following equation: [Co(NH3)5Cl]2++ OH- [Co(NH3)5(OH)]2++ ClIt involves following two mechanisms. 1. SN-2, Displacement Mechanism The reaction proceeds as: ( OH ) fast [Co(NH3)5(OH)Cl]+ [Co(NH3)5(OH)]2++ Cl[Co(NH3)5Cl]2+ slow (C.N.=6) (C.N.=7) Rate of Reaction = = The rate law is 2. (C.N.=6) K[Complex][base] K[Co(NH3)5Cl][OH-] d [Co(NH3)5Cl] = KB[Co(NH3)5Cl][OH-] dt SN-1 Displacement Mechanism: The complex which acts as a Bronsted acid is converted into its conjugate base (CB), [Co(NH3)4(NH2)Cl] + which is obtained by removing a proton H+ from the amino group present in the complex. CB is an amido complex, since it contains an amido group. SN-1 mechanism fails to explain quite a few observations: (1) 7-coordinate complexes are not very stable. (2) The value of kn is nearly 104 times higher than kA. Why should hydroxyl ions posses the exceptionally high nucleophilic activity as compared to the similar anions? (3) If a proton cannot be abstracted from N5 ligands (e.g., [Co(py)4Cl2]+ or [Co(CN)5Cl]3-), reaction rate for hydrolysis is very low. To overcome the above difficulties, an alternative mechanism is proposed by Garrick (1987). In this case the OHions abstract a proton form a ligand in N5 group giving CB of the ligand. This undergoes the dissociative mechanism as shown below: fast [(NH3)4Co(NH2)Cl]++H2O (6.1) [(NH3)5CoCl]2++OH- [(NH3)4Co(NH2)]2+ + Cl- (6.2) [(NH3)4Co(NH2)Cl]+ slow fast [(NH3)5Co(OH)]2+ [(NH3)4Co(NH2)]2++H2O (6.3) The rate determining step is the dissociation of the amido complex given in Eq.(6.2) whose concentration would depend upon the concentration of hydroxyl ions present. This is the SN1CB process. The rate law will bed = [Co(NH3)5OH] = dt K1K2[Co( NH 3 )5 Cl ][OH ] K1[ H 2O] 2 K2[ H 2O] = K[Co(NH3)5Cl][OH] where, K= K1 K 2 K 1[ H 2O]2 K 2 [ H 2O] Though it seems very unlikely that reduction in one positive charge form [Co(NH3)6]3+ to [Co(NH3)5(NH2)]2+ should increase the reaction rate enormously, it is possible that through a bonding intermediate, the stability of the 5-coordinate complex is increased (Fig. 6.5). Fig. 6.5: Stabilization of the intermediate 5-coordinate species through the resonance effects involving NH2 group. The SN1 CB mechanism does not explain the following observations. (i) The conjugate base readily dissociates and releases the ligand L; and (ii) the concentration of the conjugate base is very low due to the basic nature of the ligands, and should be present only as a very small fraction of the concentration of the complex present. Direct and Indirect Evidences in Favour of Conjugate Mechanism: Equation 6.1 requires that the reacting complex should have at least one Photonic hydrogen atom (H+) on a non-leaving ligand so that H+ may transfer to OH- to form its conjugate acid H2O and conjugate base, [Co(NH3)4(NH2)Cl]+ of [Co(NH3)5Cl]2+ which acts as an acid. Thus a complex having no proton should react with OH- much more slowly and the rate of reaction would be independent of the concentration of OH-. It is observed that the complexes like [Co(Cn)2Br] and trans [Co-(Py)4Cl2]+ which does not have N-H hydrogen undergo hydrolysis much more slowly in basic solution at a rate which is independent of [OH-] over a wide range. Thus in the absence of an acidic portion on the ligands an SN1 CB mechanism is not possible. Such complexes undergo rapid base hydrolysis supports the SN 1 CB mechanism and the acid-base properties of the complexes are more important to the rate of reaction, than the nucleophilic properties of OH. Thus both the mechanisms give the same rate laws and the same hydroxo products in aqueous solution, because water is a good cocoordinating agent. The rate of formation of [Co(en)2(NO2)Y]+ depends only on the concentration of the base, OH, not on the nature or concentration of Y-, OH- and piper; dine are used as catalysts while N3-, NO2-, SCN- ion are used as nucleophiles. In SN1CB mechanism the reactions of [Co(NH3)5Cl]2+ and OH- in aqueous solution at 25oC in presence of H2O, when H2O2 is added to the reaction mixture of [Co(NH3)5Cl]2+ and OH-, the reaction between OH- and H2O2 occurs as: OH- + H2O2 H2O + HO2Which increase the rate of base hydrolysis reaction and form peroxo products. On the other hand if the reaction occurs by an SN1CB mechanism the addition of H2O2 to the reaction mixture should reduce the rate of base hydrolysis reaction compared to OH- because of the reduction in the concentration of OH- ions. The rate of SN1CB reaction is directly proportional to the concentration of OH-. 6.3.5 Anation Reaction The reaction in which an aquo ligand (i.e. H2O molecule) from an aquo complex is replaced from the co-ordination shell by some axion. [Co(NH3)5(H2O)]3+ + X- [Co(NH3)5X]2+ + H2O Thus we find that an anation reaction is the reverse of acid hydrolysis reaction. It shows that these are bimolecular reactions with a rate which depends on the concentration of the complex and X. The same second order kinetics would be observed for a unimolecular process. slow fast [Co(NH3)5]3+ [Co(NH3)5X]2++ H2O [Co(NH3)5(H2O)]3+ Let us consider replacement of water in a species containing five nonlabile ligands such as [Co(NH3)5(H2O)]3+ , and let us reverse the experimental procedure and attempt to infer kinetic behaviour from a postulated mechanism. This is k [L5M(H2O] [L5M] + H2O 1 k [L5M] + Y [L5MY] 1 Since Y competes with solvent water for the active intermediate [L5M], the rate of formation of [L5MY] can be dependent on the concentration of Y. On the other hand, there should be some high concentration of Y at which the rate of replacement of water no longer depends on the concentration of Y. The rate of formation of [L5MY] at this concentration should be equal to the rate of formation of [L5M] and also equal to the rate of exchange of water between [L5M(H2O)] and the solvent. Thus the rate of formation of [L5M] is given by d [L5M] = k1[L5M(H2O)] - k-1[L5M][H2O] - k2 [L5M][Y] dt According to the steady-state approximation, the concentration of the very reactive [L5M] remains small and constant during the reactions (i.e. d [L5M] = 0 at the steady state). Thus, dt [L5M] = K1[[L 5 M( H 2O ] K 1[ H 2O ] K 2 [Y ] and d K K [L M( H 2O][Y ] [L5MY] = 1 2 5 dt K 1[ H 2O] K 2 [Y ] if k-1 [H2O] > k2[Y] d KK [L5MY] = 1 2 [L5M( H 2O][Y ] dt K 1 and a second-order reaction will be observed. On the other hand, if k2[Y] > k-1[H2O] d [L5MY] = k1 [L5 M( H 2O] dt giving first-order kinetics with the overall first-order constant equal to that for the dissociations of the aquo complex. 6.3.6 Reactions without Metal-Ligand Bond-Cleavage Many a times, replacement of ligand takes place without breaking a metal-ligand bond. Important examples of this fact are formation of aquocomplex, [Co(NH3)5H2O]3+, from carbon a to complex, [Co(NH3)5CO3] and nitrito complex [Co(NH3)5ONO]2+ from hydroxo or aquo-complex, [Co(NH3)5H2O]3+. The most likely path for the equation of the carbonato complexes seems to be the electrophilic attack by the proton on the O atom bonded to the metal, so that no O is found in the complex when the equation is carried out in presence of H2O (Fig. 6.6). Similarly the reaction of pentamineaquacobalt (III) with NO2- ion is explained by the sequence in Fig. 6.7. Fig. 6.6 Mechanism of substitution of carbonate group by water through electrophilic attack by H2O+. Fig. 6.7 Probable mechanism of substitution of [(NH3)5Co(OH)]2+ by nitrite NO2- through an electrophilic attack. Check Your Progress-2 Notes : (i) Write your answers in the space given below . (ii) Compare your answers with those given at the end of the unit. (a) SN-1 and SN-2 mechanism of substitution reactions differ in (i) In SN-1 rate determining step is.............................process, while that in SN-2 is .......................process. (ii) The rate determining step in SN-1 is...................molecular, while that in SN-2 is ....................... molecular. (iii) In SN-1 rate = ...................................... while that in SN-2 rate = ...................................... (b)(i) For acid hydrolysis at low pH, the rate Law is ...................................=................................................. (ii) The rate law of base hydrolysis reactions of an octahedral ammine complex, by SN-1 CB process is d = [Co(NH3)5OH] = ................................. dt = ................................... (iii) Formation of aquo-complex from a carbonato complex is an example of substitution...................... bond, and involves.............. attack on.................... 6.4 LET US SUM UP In order to convert reactions in to products it is necessary that the groups or the atoms linked in what ever manner in the reactants molecules should separate and then reunite in the form of the products. On the thermodynamic basis, Gibbs free energy for the reaction should decrease, in order to the reaction takes place, i.e. G should be negative. Since, G = H - T S, hence the possibility of conversion of reactants in to products is only when the state of disorder, and the bond energies in the products, are relatively high; H should be negative and S should be positive. Complexes are generally classified as labile and inert with reference to their reactivity. The ability of a complex to engage itself in the reactions involving the replacement of one or more ligands in the coordination sphere by other ligand is called lability of the complex. The complexes that undergo rapid substitution are termed labile; where as these with law rates of substitution are called inert. According to VBT, the inner orbital complexes (using d 2sp3 hybridisation for octahedral complication) are inert while the outer orbital complexes (using sp3d2 hybridisation) are labile. The inner orbital complexes may be labile only when they have at least one d-orbital in t2g set is vacant; e.g. [V(NH3)6]3+ ion, According to CFT, complexes with high values of CFSE are inert, while those with small values of CFSE are labile. Substitutions of ligands generally follow one of the two, SN-1 or SN-2 mechanisms. In SN-1 or dissociation mechanism, the rate determining slow step is a metal-ligand bond breaking step, since the coordination number of the complex, MX5Y (=6) is decreased to 5 in the intermediate, MX5, complexes: Y Z [MX5Y] [MX5] [MX5Z] (C.N.=6) (C.N.=5) (C.N.=6) Thus, the rate of SN-1 mechanism is first order with respect to MX5Y, i.e. the rate determining step is unimolecular. In SN-2 process the rate determining step involves a metal-ligand bond making step, with the increase of coordination number from 6 to 7: Z Y [LMX5] [L-MX5Z] [MX5Z] (C.N.=6) (C.N.=7) (C.N.=6) Thus, the rate determine step in SN-2 process in bimolecular i.e. its rate of reactions is second order; first order with respect to [MX5L] and first order with respect to Z. For SN-1 mechanism, rate = k [MX5L] For SN-2 mechanism, rate = k [MX5L][Z] The substitution reaction in which a ligand is replaced by a H2O molecule or by OH- group is known as hydrolysis reaction. The reaction is called 'acid hydrolysis' or 'aquation' when an aquo complex is formed by the replacement of a ligand by H 2O molecule while the reaction in which a hydroxo complex is formed by the replacement of a ligand by -OH group is called base-hydrolysis. Acid hydrolysis occur, in neutral and acid solutions (pH<3), while base hydrolysis occurs in basic solutions (pH>10). The rate of acid hydrolysis reaction is of first order [Co (NH3)5X]2+ + H2O [Co(NH3)5(H2O)]3+ + XAs in oqueous solutions the concentration of water is always constant, the rate law, K = K1 [Co (NH3)5X]2+ [55.5] does not indicate whether these reactions proceed by and SN-2 displacement of X by H2O or by SN-1 dissociation followed by the addition of H2O. For base hydrolysis; [Co (NH3)5Cl]2+ + -OH [Co(NH3)5(OH)]2+ + Cl- SN-2 mechanism gives rate of the reaction = K[Co(NH3)5Cl]2+ [OH-] and the rate law d [Co(NH3)5Cl] = KB[Co(NH3)5Cl][OH-]. dt Gerick proposed SN-1CB mechanism for base hydrolysis reaction. In this, the -OH ions abstract a proton from a ligand in N5 group, giving the conjugate base of the ligand. This under goes the dissociative mechanism: Fast [(NH3)4Co(NH2Cl)]+ + H2O [(NH3)5CoCl]2+ + OH- [(NH3)4Co(NH2)]2+ + Cl[(NH3)4Co(NH2)Cl]+ Slow Fast [(NH3)5Co(OH)]2+ [(NH3)4Co(NH2)]2+ + H2O Thus, the rate determining step is the dissociation of the amido group. The rate law will bed [Co(NH3)5OH] = dt K1K 2 [Co(NH3 )5 Cl][OH- ] K 1[ H 2O] 2 K 2[ H 2O] = K [Co(NH3)5Cl][OH-] The reactions involving removal of coordinated water molecule are known as 'anation' reactions: [[Co(NH3)5(H2O)]3+ + X- [Co(NH3)5X]2+ + H2O This reaction is reverse of acid hydrolysis reaction. The same second order kinetics would be observed for a unimolecular process: [Co(NH3)5]3+ [Co(NH3)5(H2O)]3+ Slow H 2O Fast X [Co(NH3)5X]2+ Many a times replacement of ligand takes place without breaking a metal-ligand bond, e.g. formation an aquo-complex from a carbonato complex. These involve the electrophilic attack by the proton on the O-atom bonded to the metal. 6.5 CHECK YOUR PROGRESS: THE KEY 1(a)(i) G should be negative. (ii) G = H - T S That is .......................should be very high i.e. H should be negative and T S should be positive. (b)(i) Labile complexes are outer orbital complexes....................inert complexes are inner orbital complexes. (ii) One orbital in t2g set: e.g. [V(NH3)6]3+ (iii) Have high values of CFSE. 2.(a)(i) Is metal-ligand bond breaking process that in SN-2 is metal-ligand bond making process. (ii) SN-1 is Unimolecular. SN-2 is bimolecular. (iii) SN-1, rate = K[MX5Y] SN-2, rate = K[MX5Y][Z] (b)(i) Rate law is - d [Co(NH3)5X] =KA[Co(NH3)5X] dt K1 K 2 [Co(NH 3 ) 5 Cl][OH - ] (ii) = K 1[ H 2 O] 2 K 2 [ H 2 O] = K [Co(NH 3 )5 Cl][OH - ] (iii) Without breaking M-L bond. Involves electrophilic attack of proton on oxygen bonded with metal.