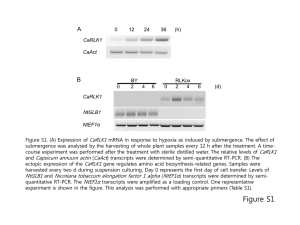
RNA-seq presentation
advertisement

Please DO NOT switch on your computers – yet. RNA-seq Analysis Graham Etherington Sainsbury Laboratory Training Course http://tsltraining.tsl.ac.uk/ Today's topics • The basics – What is RNA-seq, paired-end reads, alternative splicing • Considerations before sequencing – Library prep – What ‘contaminate’ RNA (rRNA, abundant transcripts) to remove and how. • Sequencing – Quality control – Assembly techniques • Reference-based alignment • De-novo assembly • Combined assembly (Align-then-assemble vs Assemble-then-align) • Choosing a strategy and a program • Expression analysis Today's topics • Tutorials – Reference-based transcript assembly and expression analysis without annotation using Galaxy • TopHat – Cufflinks - Cuffmerge - Cuffdiff – De-novo assembly using Trinity What is RNA-seq? Genome Genes Extract mRNA (expressed genes) Sequence mRNA Assemble into transcripts RNA-seq basics - Paired-end reads • Sequences can be paired-end – sequences occur as ‘pairs’ with one left-hand (forward) read and one right-hand (reverse) read. – a given distance (insert-size) between the start and end of pairs. Paired -ends Left (forward) read 76 nucleotides Right (reverse) read 76 nucleotides 500 nt DNA fragment ~350 nt gap ~500 nt ‘insert size’ RNA-seq basics - Alternative splicing RNA-seq – the basics • • • • • Genome of interest. How many genes (mRNAs) are there? Are some novel? Alternative spliced isoforms? Which genes are expressed under different environmental conditions (cf microarrays)? • Are some expressed more than others? Pre-sequencing • Library prep. • Multiple insert sizes captures both short and long transcripts plus alternative spliced isoforms – longer insert sizes offer long-range exon connectivity • Which RNA to select – poly-A tail RNA – misses ncRNA + rare mRNAs without poly-A tail – leave all RNAs in then remove rRNA by ‘hybridisation-based depletion methods’ • biases quantification of high-abundant transcripts • Strand-specific protocols – Aids assembly and quantification of overlapping transcripts from opposite strands Post-sequencing • Quality control • LOTS of data – don’t worry about throwing a lot of it away – remove short/long reads – remove reads with Ns – remove PCR duplicates – remove/trim low-quality reads/regions – Remove low copy k-mers Reference-based Alignment • Use when a closely-related reference is available. • 3 steps ① Use a splice-aware aligner (e.g. BLAT, TopHat). ② Cluster reads from each locus to build isoform graphs. ③ Traverse graph to resolve isoforms (e.g. Cufflinks, Scripture) Splice-aware aligners • Two types- Seed & extend and BWT Seed-and-extend SEED-part of read GGACG EXTEND alignment ATGGACGTCATGTTC Reference Splice-aware aligners • Burrow-Wheeler transform (BWT) • Creates a compressed ‘index’ of the genome. • Stretches of sequence can be ‘looked-up’ – Narrows-down the search space – Speeds up alignment – Requires less memory Creating and Traversing Graphs Reference-based Alignment • Applications: – Microbes and lower eukaryotic organisms. – Few introns and little alternative splicing – Use with strand-specific sequencing to identify overlapping genes. Reference-based Alignment • Advantages: – Contamination not a great problem – won’t align. – Less memory use – Align low-abundance transcripts – Identify transcripts undiscovered in annotated reference Reference-based Alignment • Disadvantages: – Relies on the accuracy of the reference sequence • May contain errors, deletions, missassemblies. • Can miss divergent transcripts – Reads often align to multiple regions • Excluding multi-mapped reads – leaves gaps • Randomly assign multi-mapped reads – false transcripts – Can’t easily assemble trans-spliced genes Reference-based Alignment • Summary • Preferable where a high-quality reference exists. • Can assemble full-length transcripts at depth of 10x. • Can include longer reads (e.g . 454) to capture connectivity between more exons. De-novo assembly • Doesn’t use a reference sequence. • Finds overlaps between reads and assembles them into contigs/transcripts. • Constructs De Bruijn graph which breaks reads into k-mers and connects overlapping nodes. De Bruijn graphs All substrings of length k (k-mers) are generated from each read. De Bruijn graph created by kmers that overlap by k–1. Single-nucleotide differences cause 'bubbles' of length k in the De Brujin graph Insertions or deletions introduce a shorter path in the graph. Collapse adjacent nodes. Calculate paths through graph. Isoforms. De-novo Assembly • Applications: – Microbes and lower eukaryotic organisms. – Yeast transcriptomes can be assembled with >30x coverage. – Overlapping genes from opposite strands can be detected by not allowing reverse complements in De Bruijn graph and using odd k-mers. – Higher eukaryotes more challenging due to larger datasets and difficulties in identifying alternative splice sites. De-novo Assembly • Advantages – Doesn’t need a reference sequence. – Sometimes better than reference-based assembly when: • reference is of low quality (e.g. missing bits). • Unknown exogenous transcripts want to be detected. • Where long introns are expected. – Doesn’t depend on the correct alignment of reads to splice sites. De-novo Assembly • Disadvantages: – With higher eukaryotic datasets needs lots of RAM – Requires higher sequencing depth than referencebased assembly (30x cf 10x). – Highly similar transcripts are likely to be assembled into single transcripts. – Sensitive to read-errors. Hard to tell errors from low-abundance transcripts. Combined strategy • Use both de-novo assembly and referencebased alignment methods to get the best results. • Two techniques: – Align-then-assemble – Assemble-then-align • Make use of sensitivity of reference-based aligners and use de-novo assembly for novel sequences. Combined strategy • Align-then-assemble – Most intuitive. – Align reads to a reference. – What doesn’t align – denovo assemble. Combined strategy • Assemble-then-align – When quality of reference genome is suspect. – When reference genome is from distantly related species. – De-novo assemble into contigs first. – Then use reference to extend contigs into longer transcripts. – Small errors in the reference genome don’t get propagated into the new assembly. Choosing a strategy • Factors to consider – Reference genome available? • Good quality? • Closely-related species? – Aim of project • Annotation • Identify novel transcripts • Expression analysis Choosing a splice-aware alignment program Choosing a transcript assembly program Expression analysis The more abundant an RNA, the more times it will be randomly selected for sequencing. Gene 1 Condition A Gene 1 Condition B expressed mRNA sequencing Reads Expression analysis • Use No. of mapped reads as an indicator of expression. Map reads back to genome Gene 1 Condition A Gene 1 Condition B Expression analysis • Need some way to normalise the expression data. • Fragments Per Kilobase of exon per Million fragments mapped (FPKM). • Some controversy over this approach – bias for longer transcripts. Tutorials • • • • Switch on your computers and boot into Windows. Log-in using the yellow username on your machine. Go through the tutorial sheet. There are two tasks, both using Galaxy: – Reference-based transcript assembly and expression analysis without annotation using Galaxy • TopHat – Cufflinks - Cuffmerge - Cuffdiff – De-novo transcript assembly using Trinity. • Take your time during the tutorials and make sure you understand what you are doing. • Please delete your Galaxy analysis when finished. Tutorials • Logging on to your computers: – Use the name given on the yellow sticker on your machine. – Password: Learning26 • Logging into Galaxy – Go to http://galaxy.tsl.ac.uk – machine_name@nbi.ac.uk (e.g. b26stu10@nbi.ac.uk) – Password: Learning26