LectureIV
advertisement

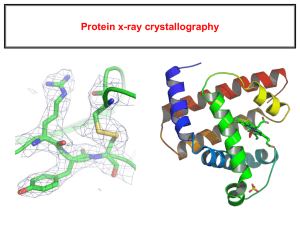
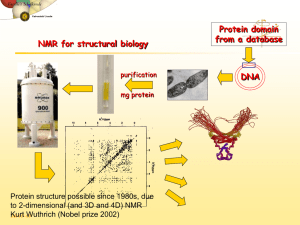
IV. Protein Structure Prediction and Determination • Methods of protein structure determination • Critical assessment of structure prediction • Homology modelling • Threading • Prediction of novel folds • Protein design Methods to determine protein structure • X-Ray and NMR methods allow to determine the structure of proteins and protein complexes • These methods are expensive and difficult – Could take several work months to process one proteins • A centralized database (PDB) contains all solved protein structures – XYZ coordinate of atoms within specified precision – ~19,000 solved structures X-ray crystallography and NMR are the two major techniques for determining protein structures X-ray X-ray crystallography: Protein isolation Crystal Protein Purification Phases of diffracted rays Protein Crystallisation Electron density Protein model X-ray crystallography Liquid nitrogen is used to freeze the crystal which allows for increased reliability of information gathered from testing. The area detector, which collects the diffracted x-rays once they pass through the crystal, is the black plate located behind the nitrogen stream, (right) sample x-ray diffraction pattern. The phase problem: Isomorphous Replacement: combination of diffraction data from the native crystal with data from other crystals containing the same protein packed in the same way but adding a heavy atom Molecular Replacement: placement of a known relative structure in different positions and orientations, providing approximate phases Multiwavelength Anomalous Dispersion: Measurements of the variation of the intensity distribution in the diffraction pattern over a range of wavelengths Direct Methods: Knowledge of electron density distributions in crystals permits calculation of phases directly from experimental data Phase determined Experimental data: Three dimentional coordinates Relative mobility of atoms Model built over it Refinement of the model comparing with empirical data Optimised protein structure X-ray crystallography Limitations • An extremely pure protein sample is needed. • The protein sample must form crystals that are relatively large without flaws. Generally the biggest problem. • Many proteins aren’t amenable to crystallization at all (i.e., proteins that do their work inside of a cell membrane). Measures of structural quality R-factor is a measure of how well the model reproduces the experimental intensity data, the lower this factor the better the structure. R = 0% There is no experimental error (ideal) R = 60% Atoms placed randomly in the crystal R 20 Good structure prediction The free R-factor is an unbiased measure of the agreement between the model and a subset of experimental data withheld during the refinement process Good protein structures: 1. Are compact as measured by their surface area and packing density 2. Have hydrogen bonds with a reasonable geometry, and with all the hydrogen bonds determined 3. Their backbone conformation angles are confined to the allowed areas of the Sasikharan-Ramakrishnan-Ramachandran diagram Nuclear Magnetic Resonance Nuclear magnetic resonance (NMR) spectra measure the energy level of the magnetic nuclei in atoms This energy depends on the effect transmitted between atoms affecting the precise frequency of the signal from an atom (chemical shift). This chemical shift can define secondary structures NMR can determine the value of conformational angles Interactions between spatially proximal atoms (< 5Å) can be used by NMR to determine the closeness of atoms in the structure (Nuclear Overhauser effect (NOE) Peaks correspond to the interaction of pairs of atoms The spectroscopists has first to correlate peaks with amino acids in the sequence (Assign the spectrum) The data generated provide a set of distance constraints and determine the secondary structure and some indications of the tertiary interactions Nuclear Magnetic Resonance • Solving an NMR structure means producing a model or set of models that manage to satisfy all known NMR distance constraints (generated by the experiment). • NMR models are often released in groups of 20-40 models because the solution to NMR structure determination is much more ambiguous than x-ray. • NMR is limited to small, soluble proteins only. Nuclear Magnetic Resonance Sample RMN spectra Spectra procession Sequential assignation Conformational restrictions 3D structure calculation Refinment Analysis NMR vs. X-ray crystallography NMR models An X-Ray liquid crystal Protein Structures • in theory, a protein structure can solved computationally • a protein folds into a 3D structure to minimizes its potential energy • the problem can be formulated as a search problem for minimum energy – – – the search space is defined by psi/phi angles of backbone and side-chain rotamers the search space is enormous even for small proteins! the number of local minima increases exponentially of the number of residues Protein Structure Prediction • ab initio folding methods – use first principles to computationally fold proteins – not practical (yet) due to its high computational complexity • Comparative modeling – Protein threading – make structure prediction through identification of “good” sequence-structure fit – Homology modeling – identification of homologous proteins through sequence alignment; structure prediction through placing residues into “corresponding” positions of homologous structure models Protein Threading • • the basic idea – placing a protein sequence onto a structural template “optimally” – assessing how good the structure is energetically key components: – a structural template database – an “energy” function for measuring quality of a placement (alignment) – an algorithm for finding an optimal placement – a capability for assessing the reliability of prediction query sequence MTYKLILNGKTKGETTTEAVDAATAEKVFQYANDNGVDGEWTYTE template set PROSPECT Predictions t49 t57 actual predicted actual predicted t68 t70 actual predicted actual predicted How and Why Threading? The idea of threading came from the observation that most of the proteins adopt one of a limited number of folds: Just 10 folds account for the 50% of similarities between protein superfamilies Rather than trying to predict the correct structure from the unlimited number of possible structures, the protein structure might have been surely determined before for other proteins In case that our protein shares obvious similarity with other protein with a known 3D structure the folding problem is trivial It is desired, however, that threading might be able to detect structural similarities that are not accompanied by any detectable similarity Algoritmos de threading. General. 1. Library of protein structures (fold library) all known structures representative subset (seq. similarity filters) structural cores with loops removed ................ 2. Binary alignment algorithm with Scoring function contact potential environments ALMVWTGH......... Instead of aligning a sequence to a sequence, align strings of descriptors that represent 3D structual features. Usual Dynamic Programming: score matrix relates two amino acids Threading Dynamic Programming: relates amino acids to environments in 3D structure 3. Method for generating models via alignments Threading Algorithms Puntuation function -Amino acids are in similar environments to those where known structures are found ALMVWTGH......... -Contact potentials -Coincidence of predicted and real secondary structures and calculation of accessibilities -Homology matrices obtained from alignment of structures - HMMs ................ - Solvatation potentials Threading algorithms Contact potential d Count pairs of each residue type at different separations Energy of interaction = -KT ln (frequency of interactions) Boltzmann principle Jones, 1992; Sippl, 1995 d Threading algorithms Sequence profiles + secundary structure Kelley et al., 2000 http://www.bmm.icnet.uk/~3dpssm threading. Examples threading. Examples threading Post-processing of the results Combining with additional information threading Post-processing of the results Filtering models De Juan et al., 2001 MAKEFGIPAAVAGTVLNVVEAGGWVTTIVSILTAVGSG GLSLLAAAGRESIKAYLKKEI KKGKRAVIAW threading Evaluation of methods I) CASP 94, 96, 98, 00, 02 EVALUATION Databases Algorithm Computer evaluation 1/3 correct fold (ali?) MODEL(S) http://PredictionCenter.llnl.gov/casp4/ PROSPECT Predictions t49 t57 actual predicted actual predicted t68 t70 actual predicted actual predicted Why engineer proteins? • 1) Engineered macromolecules could have experimental use as experimental tools, or for development and production of therapeutics • 2) During the process of said engineering, new techniques are developed which expand options available to research community as whole • 3) By approaching macromolecule as engineer, better understanding of how native molecules function (Doyle, Chem & Bio, 1998) Ligand Binding – protein flexibility “In this study, we set out to elucidate the cause for the discrepancy in affinity of a range of serine proteinase inhibitors for trypsin variants designed to be structurally equivalent to factor Xa.” (Rauh, J. Mol. Biol., 2004) Def: Ligand Any molecule that binds specifically to a receptor site of another molecule; proteins embedded in the membrane exposed to extracellular fluid.