Drug Development with Recombinant DNA Technology
advertisement

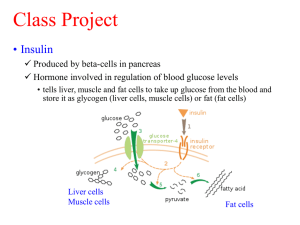
Drug Development and Evaluation Phase II May 2010 Protein-based Therapeutics Today protein therapeutics are obtained from two sources: Purified from a wide variety of different organisms or Produced by genetic engineering using recombinant DNA technology Protein-based Therapeutics Nonrecombinant proteins are purified from their native source, such as pancreatic enzymes from pig pancreas and alpha-1-proteinase inhibitor from pooled human plasma. Production systems for recombinant proteins include bacteria, yeast, insect cells, mammalian cells, and transgenic animals and plants. The system of choice can be dictated by the cost of production or the modifications of the protein (e.g., glycosylation, phosphorylation, or proteolytic cleavage) that are required for biological activity. For example, bacteria do not perform glycosylation reactions, and each of the other biological systems listed previously produces a different type or pattern of glycosylation. Protein-based Therapeutics Protein glycosylation patterns exert dramatic effects on the activity, half-life, and immunogenicity of the recombinant protein in the body. As one example, the half-life of native erythropoietin, a growth factor important in erythrocyte production, can be lengthened by increasing the glycosylation of the protein. Darbepoetin is an erythropoietin analogue engineered to contain two additional amino acids that are substrates for N-linked glycosylation reactions. When expressed in Chinese hamster ovary (CHO) cells, the analogue is synthesized with five rather than three Nlinked carbohydrate chains; this modification causes the half-life of darbepoetin to be threefold longer than that of erythropoietin. Recombinant Protein Drugs ADVANTAGES Transcription and translation of the exact human gene lead to a higher specific activity of the protein and a decreased chance of immunologic rejection by the patient ( the example of bovine insulin compared to recombinant human insulin). Recombinant proteins are often produced more efficiently and inexpensively and in potentially limitless quantity. For example, therapy for Gaucher's disease, a chronic congenital disorder of lipid metabolism caused by a deficiency of the enzyme beta-glucocerebrosidase. Most patients with this disease have an enlarged liver and spleen, increased skin pigmentation, and painful bone lesions. Although patients can be treated with beta-glucocerebrosidase purified from human placenta, this treatment requires purification of protein from 50,000 placentas per patient per year. This requirement obviously places a practical limit on the amount of purified protein available for patients with the disease. A recombinant form of beta-glucocerebrosidase is now available. Recombinant Protein Drugs ADVANTAGES (cont.) The recombinant protein eliminates the risk of transmissible (e.g., viral or prion) diseases associated with purifying the protein from human placentas. Recombinant technology allows the modification of a protein to improve function or specificity. For example, recombinant beta-glucocerebrosidase provides an interesting example. When this protein is made recombinantly, a change of amino acid arginine-495 to histidine allows the addition of mannose residues to the protein. The mannose is recognized by endocytic carbohydrate receptors on macrophages and many other cell types, allowing the enzyme to enter these cells more efficiently and to cleave the intracellular lipid that has accumulated in pathologic amounts. This results in an improved therapeutic outcome. Recombinant Protein Drugs in Medical Use erythropoietin, a protein hormone secreted by the kidney that stimulates erythrocyte production in the bone marrow. In patients with chemotherapy-induced anemia or myelodysplastic syndrome, recombinant erythropoietin is used to increase erythrocyte production and thereby improve the anemia. In patients with chronic kidney disease, whose levels of endogenous erythropoietin are below normal, recombinant protein is administered to correct this deficiency. Recombinant Protein Drugs in Medical Use Darbepoetin alfa is a recombinant variant of erythropoietin with a longer half-life. Protein glycosylation patterns exert dramatic effects on the activity, half-life, and immunogenicity of the recombinant protein in the body. As one example, the half-life of native erythropoietin, a growth factor important in erythrocyte production can be extended by increasing the glycosylation of the protein. Darbepoetin is an erythropoietin analogue engineered to contain two additional amino acids that are substrates for N-linked glycosylation reactions. When expressed in Chinese hamster ovary (CHO) cells, the analogue is synthesized with five rather than three N-linked carbohydrate chains; this modification causes the half-life of darbepoetin to be threefold longer than that of erythropoietin. EPO Darbepoetin Darbepoetin alfa has: Two additional sialic acid–containing carbohydrates (red) Up to 8 additional sialic acids Increased molecular weight (~37,100 daltons) Classes of Approved Recombinant Protein Drugs Ref: Scientific and Legal Viability of Follow-on Protein Drugs. David M Dudzinski, Aaron S Kesselheim. The New England Journal of Medicine Vol. 358, Iss. 8; pg. 843, 2008. Recombinant Protein Drugs In 2006, the recombinant proteins darbepoetin alfa (Aranesp), epoetin alfa (Epogen, Procrit), and etanercept (Enbrel) were among the year's top 10-selling pharmaceuticals on the basis of dollar value and accounted for almost 4% of the $275 billion annual U.S. pharmaceutical market. Recombinant Protein Drugs in Medical Use INSULIN insulin in the treatment of diabetes mellitus type I (DM-I) and type II (DM-II). Untreated, DM-I is a disease that leads to severe wasting and death due to lack of the protein hormone, insulin, which signals cells to perform a number of functions related to glucose homeostasis and intermediary metabolism. In 1922, insulin was first purified from bovine and porcine (pig) pancreas and used as a lifesaving daily injection in patients with DM-I. At least three challenges prevented widespread use of this protein therapy: (1) requirement of big number of animal pancreas for purification of insulin; (2 the cost of insulin purification from animal pancreas was high; (3) existing immunologic reactions of some patients to animal insulin. Recombinant Protein Drugs in Medical Use INSULIN (cont.) These problems were addressed by isolating the human insulin gene, “recombining” the gene with bacterial DNA, and engineering Escherichia coli using this recombinant DNA technology to express human insulin. By growing vast quantities of these bacteria, large-scale production of human insulin was achieved. The resulting insulin was abundant, inexpensive, of low immunogenicity, and free from other animal pancreatic substances. Recombinant insulin was the first commercially available recombinant protein therapeutic; it was approved by the US FDA in 1982, and has been the major therapy for DM-I (and a major therapy for DM-II) ever since. The 25 years since the approval of insulin by the FDA have seen a remarkable expansion of proteins in the pharmacologic therapy used by physicians to treat disease. Currently, more than 70 different proteins (over 40 of which are produced recombinantly) are approved by the FDA for clinical use. Protein-based therapies are, and will continue to be, a mainstay in treating human disease. ABD’de Bir İlacın Geliştirilme Süreci In vitro Çalışmalar Pazarlama dönemi NDA Klinik İlaç Geliştirme Çalışmaları Hayvan Çalışmaları Biyolojik Ürünler FAZ I F D A İ N C E L E M E S İ çalışmaları Etkililik Seçicilik Seçilen Toksisite Molekül Etki Mekanizması FAZ II çalışmaları FAZ III çalışmaları Kimyasal Sentez Ürünleri 0 YILLAR 2 4 IND Başvurusu (Araştırılacak Yeni İlaç Başvurusu) 5000 molekül 5 molekül 8 NDA FAZ IV çalışmaları 9 (FDA Onayı) 20 (Patent Bitimi) (Yeni İlaç Başvurusu) 1 molekül Ref: Goodman and Gilman’s, The Pharmacological Basis of Therapeutics, Mc GrawHill, 1996. Katzung, B.G., Basic and Clinical Pharmacology, Appleton & Lange, 1998 FDA report: From Test Tube to Patient 1999. May 2004 Drug Development Process Clinical Drug Testing in Humans NUMBER OF SUBJECTS LENGTH OF PHASE PURPOSE Phase I 20–100 Several months Mainly safety Phase II Up to several hundred Several months to 2 years Effectiveness, dosage and shortterm safety Phase III Several hundred to several thousand 1–4 years Safety, dosage, effectiveness Drug Development Process Phase I Studies Phase I studies generally involve between 20 and 100 healthy normal subjects and are intended to establish the safety and tolerability of a drug. If high levels of toxicity are expected, such as with many cancer drugs, patients with the target condition may be used in place of healthy volunteers. The focus of phase I investigation is the drug's overall effect and kinetics in the body, including maximum tolerated dose, absorption, distribution, metabolism, and excretion. To determine the effect of varying doses, subjects are started on a dose anticipated to have little effect, and receive increasing doses thereafter. Drug Development Process Phase I Studies (cont.) The primary goal of phase I studies is to establish safety, toxicity, kinetics, and major adverse effects. Phase I studies may involve nonblinded trials, in which subject and investigator are both aware of what is being administered. Phase I studies must yield sufficient information about a drug's pharmacokinetics to inform the design of scientifically valid phase II studies. For example, knowing the drug's volume of distribution and clearance enables study designers to determine an appropriate maintenance dose and dosing frequency for phase II and III trials. Drug Development Process Phase II Studies Phase II studies may involve up to several hundred patients with the medical condition of interest. Phase II clinical trials have multiple objectives, including the acquisition of preliminary data regarding the effectiveness of the drug for treatment of a particular condition. Like phase I trials, phase II trials continue to monitor safety. Because phase II studies enroll more patients, they are capable of detecting less common adverse events. Phase II studies also evaluate dose-response and dosing regimens, which are critically important in establishing the optimum dose or doses and frequency of administration of the drug. The trial usually compares several dosing regimens to obtain optimum dose range and toxicity information. The results of phase II studies are critically important in establishing a specific protocol for phase III studies. Drug Development Process Phase III Studies Phase III studies involve several hundred to several thousand patients and are conducted at multiple sites and in settings similar to those in which the drug would ultimately be used. Safety, dosage and efficacy properties obtained from Phase II study is tested in a larger patient population. Because of the large number of patients under study, phase III trials typically provide an adequate basis for extrapolating the results to the general population.