Overview_of_drug_development_cmh_with_animatiions
advertisement

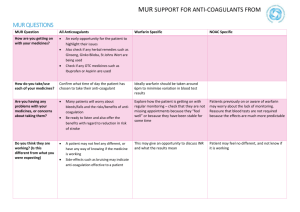
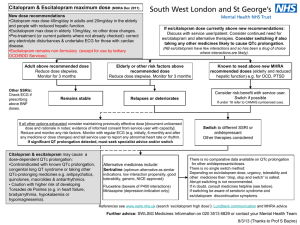
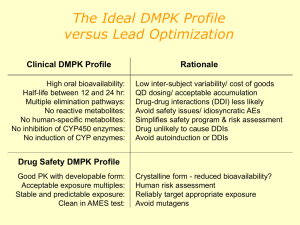
Development of a Medicine Dr Ciara Hughes January 2011 DISCOVERY OF NEW MEDICINES Time and Money Discovery of new chemical entities (NCE’s) Preclinical development Clinical development ( I-IV) http://www.wellcome.ac.uk/Education-resources/Teaching-and-education/BigPicture/All-issues/Drug-development/WTX042562.htm WHAT IS A DRUG? “Any chemical agent that affects the processes of living” A medicinal substance that interacts with a biological system - and changes it Those operating within the herbal medicine sector need to understand and comply with the Traditional Herbal Medicines Registration Scheme (THMRS), as required by Directive 2004/24/EC on Traditional Herbal Medicinal Products. Other examples Morphine from the Poppy plant Digoxin from the Foxglove Aspirin from apple skin Taxol from the Yew tree The cost of drug development Costs of drug development £500 million for one drug Takes 15-16 year to develop a drug 75% of this cost is attributable to failure 90% of all drugs developed don’t make it to the market Allocation of funds Allocation of time (in years) PRECLINICAL DEVELOPMENT Identification of drug targets Identification of drug targets Identification of drug targets Identification of drug targets Identification of drug targets Identification of drug targets Identification of drug targets Identification of drug targets Biological activity Biological activity Preclinical Information Required Before A Selected Drug Candidate Can Be Administered To Humans Pharmacological activity Pharmacokinetics and drug metabolism Toxicology Pharmaceutics EVIDENCE OF PRIMARY PHARMACOLOGICAL ACTIVITY Decision to proceed to man only after thorough characterisation of dose/concentration response relationships of candidate compound in vitro and in vivo This is the scientific basis for all rational drug development today and the essential information required before first pharmacological studies in man Behavioural Activity and CNS (including dependence) General observational studies in animals. CNS evaluation. Psychomotor testing. PHARMACOKINETICS Absorption Distribution Metabolism Excretion PHARMACOKINETICS AND DRUG METABOLISM ► PK of drug in rodents, dogs and primates as predictor of the human situation ► PK in animals is an essential part of drug selection to define the desired kinetic profile for a drug eg. half-life and bioavailability Toxicity Package Generated Before Phase 1 Trial Safety pharmacology – CVS, CNS, RS PK’s – preliminary studies on ADME Acute toxicity – 2 species by 2 routes of administration; evaluation of MRD Repeat dose toxicity – rodent and non-rodent; two 14 day studies before man Reproductive toxicology – embryo/foetal development studies – 2 species Mutagenicity/chromosomal PHARMACEUTICS Provide pharmacist with information for optimal formulation Concentrations and volumes of injections Strength of capsules or tablets Choose unit doses providing greatest flexibility Phase I Initial studies to evaluate Pk, Pd, tolerability. 30- 50 volunteers usually healthy. Starting dose. Single Ascending Dose (SAD). Multiple Ascending Dose (MAD). 80% of drugs fail at phase 1 trials OBJECTIVES OF THE FIRST STUDY Define success/fail criteria of the drug before starting the study Study objectives based on a combination of: -Tolerability/ “safety” - Kinetics - +/- Dynamics - +/- Quantitative dose-response, concentration-response relationships FACTORS TO BE CONSIDERED IN DECIDING THE STARTING DOSE Maximum no effect dose/exposure in toxicity studies using most sensitive species (NOAEL) Nature & severity of toxicity seen in animals Nature of PD activity in animals and slope of dose-response curve PK REASONS FOR STOPPING DRUG DEVELOPMENT Half-life (t ½) too short or too long Poor bioavailability Inconsistent bioavailability with low therapeutic index Saturable clearance mechanisms Multiple metabolites not covered by toxicity studies FINDINGS LIKELY TO LEAD TO PROJECT TERMINATION Poor tolerability at therapeutic concentrations Unsatisfactory kinetics/metabolism Low potency Absence of efficacy Phase II Small scale studies in patient population. Pharmacokinetics. Pharmacodynamics / surrogate endpoints Tolerability. 250-500 human exposures. Decisions before full Phase 3. Phase III Large scale efficacy trials (1000s patients). Comparator vs placebo. Several hundred to several thousand. Power curves. Ongoing PK studies special populations. Ongoing formulation studies. Ongoing toxicity / oncogenicity studies. Usually 2 successful phase 3 trials required for FDA approval Phase IV Post licensing safety evaluation Black triangle ► 1: Black triangle, spontaneous reporting ► 2: Periodic drug safety reporting Pharmacovigilance (PV) Safety Assessment of Marketed Medicines What is pharmacovigilance? Pharmacovigilance is the science of collecting, monitoring, researching, assessing and evaluating information from healthcare providers and patients on the adverse effects of medicines, biological products, herbals and traditional medicines with a view to: Identifying information about potential new hazards Preventing harm to patients. Legislation 2004 Medicines for Human Use Act The MHRA Pharmacovigilance Inspectorate is part of the Inspections and Standards Division of the MHRA. It assesses pharmaceutical companies’ compliance with UK and EU legislation relating to the monitoring of the safety of medicines given to patients http://www.youtube.com/watch?v=wvDvAEmq-cM